

 With its introduction at the HUPO World Congress in 2010 in Sydney Australia by Ruedi Aebersold, SWATH® Acquisition instantly intrigued scientists around the world. Here was a new technique with the potential to revolutionize the way proteomics studies were performed! Based on a data independent acquisition strategy using a SCIEX TripleTOF® 5600 system, SWATH was able to consistently identify and quantify at least as many peptides and proteins as other far more mature proteomics strategies on the market, but with quantitative accuracy and reproducibility rivaling gold standard MRM experiments! This solution was made broadly available to researchers with a full launch of SWATH Acquisition in the Analyst® TF 1.6 Software on the TripleTOF 5600+ System at ASMS 2012 in Vancouver (A Mine of Quantitative Proteomic Information. Prof Dr. Ruedi Aebersold, Head of the Department of Biology, ETH Zurich).
With its introduction at the HUPO World Congress in 2010 in Sydney Australia by Ruedi Aebersold, SWATH® Acquisition instantly intrigued scientists around the world. Here was a new technique with the potential to revolutionize the way proteomics studies were performed! Based on a data independent acquisition strategy using a SCIEX TripleTOF® 5600 system, SWATH was able to consistently identify and quantify at least as many peptides and proteins as other far more mature proteomics strategies on the market, but with quantitative accuracy and reproducibility rivaling gold standard MRM experiments! This solution was made broadly available to researchers with a full launch of SWATH Acquisition in the Analyst® TF 1.6 Software on the TripleTOF 5600+ System at ASMS 2012 in Vancouver (A Mine of Quantitative Proteomic Information. Prof Dr. Ruedi Aebersold, Head of the Department of Biology, ETH Zurich).
As a new technique, there were lots of different areas to investigate to see if the technique could be even further optimized. One area of investigation was the window width used for precursor ion selection. Using SWATH, every detectable precursor is sequentially fragmented by stepping a precursor isolation window of defined width across the entire mass range. Wider width windows enable fast scanning and shorter cycle times. Conversely, narrow width windows provide higher specificity and higher quality quantitation. These two conflicting requirements prompted the innovation of variable width windows.
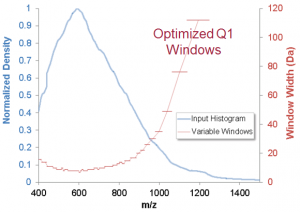 With variable width Q1 isolation windows, the width of the precursor window is adjusted to match the complexity of the precursor ion region. Narrower windows are used where more precursors are detected and wider windows where there are fewer precursors, providing a constant complexity of precursors to be maintained within the selection. This smart refinement in using SWATH prompted a beta program for Variable Window SWATH Acquisition in late 2013 to the participants in the SWATH Acquisition Multi-laboratory assessment project. Shortly after that, Variable Window SWATH Acquisition was launched as part of Analyst TF 1.7 at ASMS 2014 in Baltimore along with the TripleTOF 6600 System. At this point, researchers had increased the total number of windows from the original 30 to 60 windows. It was then realized that the sensitivity and speed of the TripleTOF platform could allow us to run up to even 100 variable windows without a decay in quantitative quality.
With variable width Q1 isolation windows, the width of the precursor window is adjusted to match the complexity of the precursor ion region. Narrower windows are used where more precursors are detected and wider windows where there are fewer precursors, providing a constant complexity of precursors to be maintained within the selection. This smart refinement in using SWATH prompted a beta program for Variable Window SWATH Acquisition in late 2013 to the participants in the SWATH Acquisition Multi-laboratory assessment project. Shortly after that, Variable Window SWATH Acquisition was launched as part of Analyst TF 1.7 at ASMS 2014 in Baltimore along with the TripleTOF 6600 System. At this point, researchers had increased the total number of windows from the original 30 to 60 windows. It was then realized that the sensitivity and speed of the TripleTOF platform could allow us to run up to even 100 variable windows without a decay in quantitative quality.
Next, there came the recognition that there was more information to be pulled out of these data-rich data files and that using larger and larger ion libraries for data processing could further increase the number of peptides and proteins quantified. Investigations showed that steady improvements in results could be observed as the library size became larger. In particular, when many variable windows were used, the larger ion libraries were able to fully leverage the added data quality and information content in the data file (Want More Protein Coverage? Use a Bigger SWATH Library).
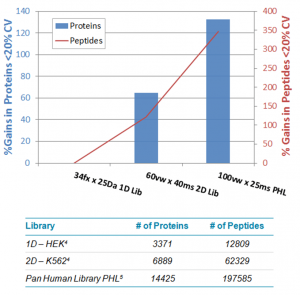 To benchmark how these innovations have impacted results quality, we did a short study where we investigated the series of method improvements back to back on the same instrument and sample. The use of variable windows and optimized ion libraries provided tremendous gains in results of this particular study. As shown below, a 300% gain in the number of peptides and a 120% gain in the number of proteins quantified were observed across replicate studies. Read the full study.
To benchmark how these innovations have impacted results quality, we did a short study where we investigated the series of method improvements back to back on the same instrument and sample. The use of variable windows and optimized ion libraries provided tremendous gains in results of this particular study. As shown below, a 300% gain in the number of peptides and a 120% gain in the number of proteins quantified were observed across replicate studies. Read the full study.
With the amount of data that was now being generated with SWATH Acquisition, it became obvious that there was a need for a more efficient and powerful processing protocol. To answer that call, the OneOmics™ project was initiated, and a beta program was rolled out at HUPO in Madrid 2014. The OneOmics project streamlined proteomics SWATH data processing with automated tools and a cloud-based platform, allowing scientists to share results easily and to use additional tools to perform meta-analysis with genomic data and pathway investigations (Industrialize your Quantitative Proteomics – Cloud Computing. Cloud Computing for SWATH Acquisition using the OneOmics™ Project).
With the back-end data processing well on its way to being optimized, now the constraint was moved to the front end. Typically proteomics experiments are performed using nanoflow chromatography to provide as much sensitivity as possible. But nanoflow chromatography can be extremely slow and requires some expertise to use routinely. The proteomics community needed techniques that could handle higher sample loads and faster turn-around times. Microflow LC coupled with SWATH Acquisition was found to be a robust solution with minimal compromises in sensitivity for performing higher throughput quantitative proteomics and enabling the analysis of hundreds of similar samples reliably and with moderate effort. In early 2016, Microflow SWATH Acquisition was introduced as a robust solution ideal for the larger cohort studies that researchers are now analyzing, providing 1 hour run-time per sample that can allow for processing of ~150 proteomes per week with excellent quantitative reproducibility and workflow robustness (Industrialize your Quantitative Proteomics – Microflow SWATH: High Throughput, Robust, Sample Analysis With Microflow SWATH® Acquisition).
The demand for SWATH Acquisition was reaching experienced proteomics researchers as well as scientists new to the proteomics workflows. This prompted the development of an easy-to-use kit to help users get up and running fast with SWATH Acquisition and to easily benchmark their entire system once established. The SWATH Acquisition Performance Kit was introduced at ASMS in 2016 and allows scientists to set-up and optimize each system component and then assess the performance of their entire SWATH workflow using standards that mimic real-world samples. The kit helps to ensure consistent performance over long periods of time, with many samples, multiple users, and multiple laboratories and facilitates switching between nanoflow and microflow.
Whether the need is for best-in-class results, data completeness, reproducibility, data re-mining, or industrialized proteomics, innovations provided over the last 5 years have enabled the SWATH Acquisition technique to be embraced by scientists around the world.
But we’re not finished yet. Look for further innovations from the SCIEX team in the future as we continue to listen to and to innovate our technologies to meet your needs.
Happy Birthday to SWATH Acquisition!



 Contact Support
Contact Support
0 Comments