

 Cloud Computing for SWATH® Acquisition using the OneOmics™ Project
Cloud Computing for SWATH® Acquisition using the OneOmics™ Project
Part 2 of a 3-Part Blog Series
For many labs, the days are long gone when it was acceptable to run only a few samples a week for your quantitative proteomics projects. The pressure for faster turn-around times, to support larger cohort studies, to sustain multiple research directions, and to transition from a purely unbiased discovery mode to verifying something truly unique and interesting, all demand a faster pace. Many labs are now being asked to analyze a hundred samples a week or more. In part 1 of this blog series, we saw how moving to a microflow SWATH workflow can dramatically increase your throughput with little compromise on overall results. In this part, we’ll address what to do with all of this data because it’s just no good if all we’ve done is move the bottleneck downstream.
We can fix that.
You’ve industrialized your sample analysis. Now it’s time to industrialize your data processing. Cloud computing with easy-to-use applications can deal with this onslaught of data by providing massive speed and scale as well as a suite of powerful processing tools that can quickly turn data bytes into biological answers. The OneOmics™ Project is a cloud computing effort that offers a suite of SWATH data processing tools with the speed, scale, and security to support even very large cohort studies. With cloud computing and the apps in OneOmics, SWATH data can be processed ~10x faster than on the desktop solution. And now with the new AutoUploader feature in CloudConnect, data files are moved automatically to the cloud right after acquisition to further streamline your quantitative proteomics workflow.
As shown in the figure below, once the SWATH data files are automatically uploaded, peptide and protein data is extracted and scored, metadata assigned, and data quality can be assessed with the Analytics application. From here, the data can be normalized across experimental groups and the protein expression differences determined using the Assembler application. The Browser application and iPathwayGuide applications provide various ways to evaluate the data.
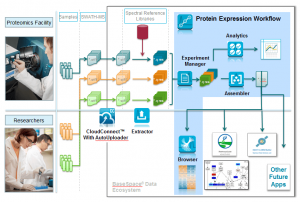
The full suite of the OneOmics application from SCIEX is now fully validated so you can have full confidence in the results! In the latest Protein Expression Workflow, we have added context sensitive help, improved the usability and visualizations and added figure exports for easy publication, all in response to user feedback. Also, principal component analysis (PCA) and principle component variable grouping (PCVG) have been added to Browser to provide even more informative visuals.
Dr. David Boocock from Nottingham Trent University shows how he uses the tools for a very interesting project on the study of the epithelial to mesenchymal transition (EMT) in prostate cancer2. The lab has developed and characterized a number of cell lines with a range of EMT behavior and then analyzed these with microflow SWATH Acquisition. Rapid analysis using the informative tools in OneOmics allowed the team to quickly validate all the biological characterization results at the proteome level.
Dr. Katy Williams from UCSF describes a pilot study where they investigated the value of using multi-omics for the study of placental development3. SWATH Acquisition on the TripleTOF® 6600 System was used for differential protein expression analysis on the samples, and RNA-Seq (HiSeq, Illumina) was used for quantitative transcript abundance. Proteomic and transcriptomic quantitative data was combined using the iPathwayGuide Software (Advaita) to investigate the implicated pathways and biological processes.
 With larger datasets comes more workflows with independent processing requirements and the full suite of OneOmics continues to grow. One of the newest applications to be added can convert any proteins of interest found in SWATH results into an MRM assay. The SWATH to MRM Builder3 application selects differentially expressed proteins (or user submitted protein list), extracts key information (best peptides, Q1 and Q3 information for each, and retention times), then outputs a text file that can be imported into Skyline Software for final assay refinement. It’s now easier than ever to bridge the gap between discovery and targeted proteomics experiments!
With larger datasets comes more workflows with independent processing requirements and the full suite of OneOmics continues to grow. One of the newest applications to be added can convert any proteins of interest found in SWATH results into an MRM assay. The SWATH to MRM Builder3 application selects differentially expressed proteins (or user submitted protein list), extracts key information (best peptides, Q1 and Q3 information for each, and retention times), then outputs a text file that can be imported into Skyline Software for final assay refinement. It’s now easier than ever to bridge the gap between discovery and targeted proteomics experiments!
 The OpenSWATH software for extracting SWATH datasets has been made available as an application within BaseSpace by from Dr. Aebersold’s team at ETH Zurich. This provides user-friendly access to this powerful software and data from this feeds into the current OneOmics applications. Stay tuned for more information on how to get starting using OpenSWATH!
The OpenSWATH software for extracting SWATH datasets has been made available as an application within BaseSpace by from Dr. Aebersold’s team at ETH Zurich. This provides user-friendly access to this powerful software and data from this feeds into the current OneOmics applications. Stay tuned for more information on how to get starting using OpenSWATH!
To learn more about OneOmics, how to use BaseSpace applications, and ask questions, see the Cloud Talk Series. To learn more about how you can improve your efficiency and output for your quantitative proteomics sample acquisition, see part 1 of this blog series and part 3.
References
- AutoUpload Cloud Talk
- A Novel Spontaneous EMT Model of Prostate Cancer to Understand the Molecular Mechanism of Epithelial to Mesenchymal Transition – Dr. David J Boocock, Nottingham Trent University
- Investigating Cytotrophoblasts in Placental Development – Using OneOmics and iPathwayGuide for MultiOmics Research – Dr. Katy Williams, University of California, San Francisco
- SWATH Acquisition to MRM Builder App Cloud Talk



 Contact Support
Contact Support
0 Comments