Abstract
Prime editing (PE) is a novel variation on CRISPR systems utilizing a fused Cas9 protein and a pegRNA to achieve high editing efficiency and precision.1 However, the length requirement of pegRNAs at 120–250 nucleotides (nt) and the potential secondary structures due to complementary or partially complementary sequences present analytical quality control (QC) challenges for purity analysis of chemically synthesized pegRNA molecules. Here, a capillary gel electrophoresis (CGE) based method was developed to mitigate the technical challenges by thoroughly disrupting the hydrogen bonds in pegRNA molecules and maintaining their denatured state during analysis. This method can offer high resolution and provide purity quantification of these pegRNAs.
Introduction
PE is a gene-editing strategy that can perform targeted, small insertions, deletions, and substitutions without introducing double- stranded breaks in the genome.1,2 The pegRNAs utilize a novel Cas9 reverse transcriptase fusion protein, guide the protein to the targeted location in the genome and provide a site-specific DNA repair template for reverse transcriptase priming.3 This technology has the advantages of high precision, efficiency and flexibility over conventional genome-editing approaches, and it shows the potential to correct genetic variants associated with human diseases.3
In some PE applications, synthetic RNA oligonucleotides are favored because they allow for the addition of stabilizing chemical modifications, and they are readily amenable to high-throughput screening, which is essential as pegRNA design rules are still being understood. However, the length requirement of 120–250 nucleotides (nt) and the complexity of pegRNAs present quality control (QC) challenges in analytical method development. The high-level secondary structures of pegRNAs can lead to shifts of migration times for pegRNAs that do not resolve in a size- dependent manner. Using common denaturants alone in the analytical analysis will not entirely disrupt the hydrogen bonds of these molecules or maintain the denatured state, resulting in inaccurate determination of the purity of the chemically synthesized pegRNAs. Therefore, due to diverse research and therapeutic interest in PE, there is a need to develop purity analysis methods that can overcome the formation of energetically favorable secondary structures of these chemically synthesized RNA molecules.
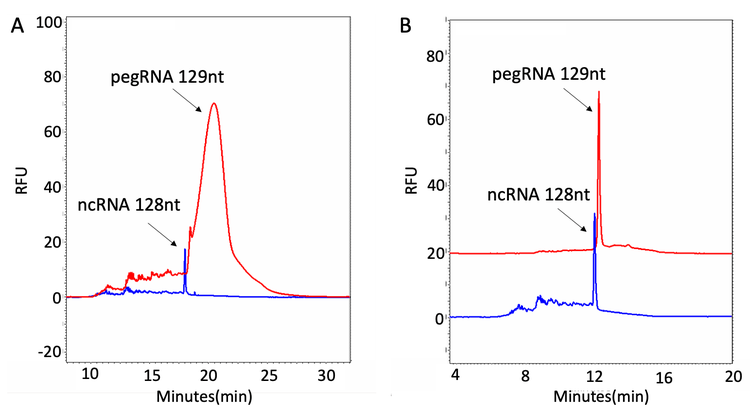
This technical note outlines a robust method that uses capillary gel electrophoresis (CGE) to mitigate the technical difficulties involved in the purity analysis of pegRNAs with high-level secondary structures (Figure 13). When synthetic pegRNAs are analyzed on a capillary electrophoresis (CE) system under standard conditions, these secondary structures produce peaks with higher structure isoforms that significantly interfere with the purity analysis (panel A in Figure 1). However, with optimized denaturing strategies, fast and reliable purity determination of pegRNA has been achieved (panel B in Figure 1).
Key features of pegRNA purity analysis
-
Innovative purity analysis workflow for chemically synthesized pegRNAs with high-level secondary structures
-
Purity determination of pegRNA from the high-resolution separation of the full-length product from its impurities
-
A high level of repeatability with relative standard deviation% (RSD%) of <3% was achieved as a result of accurate and stable capillary temperature control on the PA 800 Plus system
-
Fast quality assessment of chemically synthesized pegRNAs within 30 minutes per sample
Materials and methods
Materials: The Sample Loading Solution (SLS, PN 608082) and the ssDNA 100-R kit (PN 477480) containing DNA capillaries, ssDNA 100-R gel (lyophilized), Tris-Borate buffer, 7M Urea, and pd(A) 40-60 test mix were from SCIEX (Framingham, MA). Nuclease-free water (PN AM9932) and SYBR Green II RNA gel stain, 10,000X concentrate in DMSO (PN S7586) were from Thermo Fisher Scientific (Waltham, MA). Recombinant Human RNA Helicase A protein (PN ab114300) was from Abcam (Cambridge, UK). The single guide RNAs (sgRNAs) and pegRNA oligonucleotides were chemically synthesized and provided by Integrated DNA Technologies (Coralville, IA).
Instruments and software: A PA 800 Plus system equipped with a laser-induced fluorescence (LIF) detector and an ultraviolet (UV) detector was from SCIEX. The excitation wavelength was at 488 nm, and the emission wavelength was at 520 nm when using the LIF detector. The wavelength was 254 nm when using a UV detector. The nanoVials (PN 5043467) from SCIEX were used for sample loading. Data acquisition and analysis were performed using 32 Karat software version 10.
Methods
Tris-Borate-Urea buffer reconstitution: A total of 135 mL of nuclease-free water was added to the bottle containing the dry Tris-Borate buffer. The buffer solution was stirred for about 20–30 minutes using a magnetic stir bar until the solid was completely dissolved. The dry 7M Urea was then slowly added to the Tris- Borate buffer. The solution was continuously stirred for up to 2 hours at room temperature until the 7M Urea was completely dissolved. The reconstituted Tris-Borate-Urea buffer should be good for 30 days if stored at 2°C to 8°C after preparation. The entire bottle of buffer should be brought to ambient temperature before use. The required volume to be used for the day was removed and filtered through a 0.2 μm disposable syringe filter into a clean container.
ssDNA 100-R gel buffer reconstitution: A certain amount (varied in the method optimization) of 0.2 μm filtered Tris-Borate- Urea buffer was added to the lyophilized gel to reconstitute the gel buffer. After the gel buffer vial was securely capped, the gel mixture was gently stirred with a stirring bar for 4 to 6 hours or overnight until the gel was completely dissolved. The gel buffer was then filtered through a 0.45 μm disposable syringe filter. The prepared gel buffer can be used for up to 30 days after preparation if stored at 2°C to 8°C.
Cartridge assembly: A DNA capillary (PN 477477) from SCIEX was installed according to the instructions in the ssDNA 100-R kit application guide.4 The total capillary length was 30.2 cm, with 20 cm as the length from the detection window. A 100 x 200 μm aperture was used for better resolution when using a UV detector. Since the inner wall of the DNA capillary is coated, the cartridge assembly should be carried out promptly. Excessive exposure to air may damage the inner coating and cause clogging. The capillary ends must be immersed in liquid (water or buffer) as soon as the cartridge assembly is complete to prevent the coating from drying out.
Preparation of buffer trays and sample trays: Vial positions for buffer trays are indicated in Figure 2. Each water vial was filled with 1.5 mL double deionized (DDI) water. The waste vial was filled with 0.8–1.0 mL DDI water. The gel vial was filled with 1.3 mL ssDNA 100-R gel buffer. The gel vial was capped and sonicated to remove the air bubbles before being loaded onto the PA 800 Plus system for analysis. Specifically, the gel vial was sonicated five times for 30 seconds each time, allowing the air bubbles to rise to the surface after each 30-second interval. The buffer vials were filled with 1.5 mL Tris-Borate-Urea buffer.
Instrument setup: The Initial Conditions and UV Detector Initial Conditions were set up as indicated in Figure 3 and Figure 4, respectively. The same setup was used for all three methods: the new capillary-conditioning method, the gel-filling method and the separation method.
Gel filling and separation: The Time Program settings for the new capillary-conditioning method are shown in Figure 5. Figure 6 and Figure 7 show the Time Program settings for the gel-filling and separation methods, respectively.
Sample preparation: The RNA oligonucleotides were diluted with nuclease-free water to about 0.5–1 μM and then loaded into the sample insert tube (a minimum sample volume of 40 μL) or the nanoVial (a minimum sample volume of 5 μL). Sample vials were loaded onto the sample inlet tray for analysis.
To analyze oligonucleotides with ultra-low concentration, the SYBR Green II RNA gel stain and the LIF detector were used to achieve high sensitivity. First, the Tris-Borate-Urea buffer was mixed with the SYBR Green II RNA gel stain at a ratio of 25,000 to 1. The settings for the LIF Detector Initial Conditions are shown in Figure 8.






Results and discussion

Workflow of oligonucleotide purity analysis with single-base resolution
The workflow of oligonucleotide purity analysis shown in Figure 9 utilizes the ssDNA 100-R kit and the PA 800 Plus system with simple dilute-and-load sample preparation. The ssDNA 100-R kit is suitable for both small-sized RNA and DNA purity analysis. It features a replaceable gel buffer and a coated capillary for maximum reproducibility, and it provides rapid separation and analysis of oligonucleotides between 10 and 100 bases with single-base resolution. The results shown on the right side of Figure 9 demonstrate the separation of a poly d(A) 40-60-mer oligonucleotides test mix with single-base resolution (upper panel) and the separation of the 1:1 mixture of a 99 nt and 100 nt DNA oligonucleotide (lower panel).
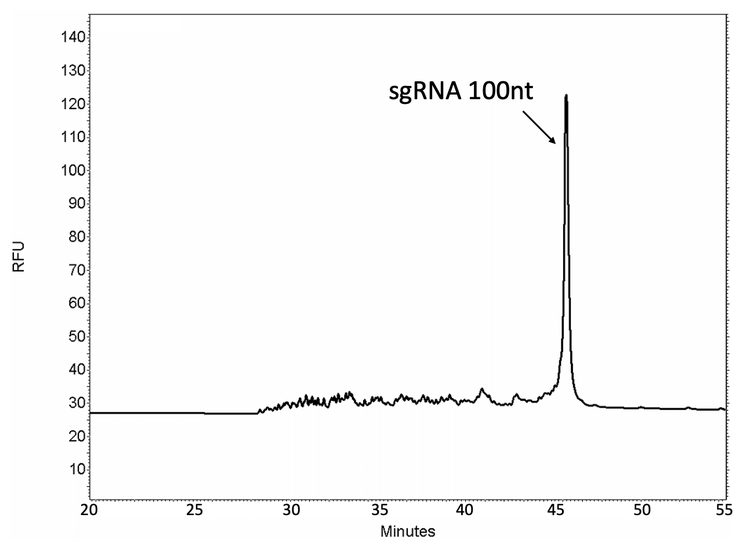
The purity analysis of a 100-nt single guide RNA (sgRNA) is shown in Figure 10. The purity of the synthesized product can be accurately determined by separating the full-length product (FLP) from its lower molecular weight in-process impurities.
Method development for purity analysis of pegRNAs
The gel matrix in the ssDNA 100-R kit, which is suitable for oligonucleotide analysis up to 100 nt, needs to be optimized for pegRNA analysis due to the larger length requirement (120–250 nt) for pegRNAs. Therefore, using 10 mL of 0.2 μm filtered Tris- Borate-Urea buffer for reconstitution of the lyophilized gel has been selected to provide the optimal resolution and separation time for the RNA ladder in the 100 nt to 250 nt range (data not shown in this technical note).
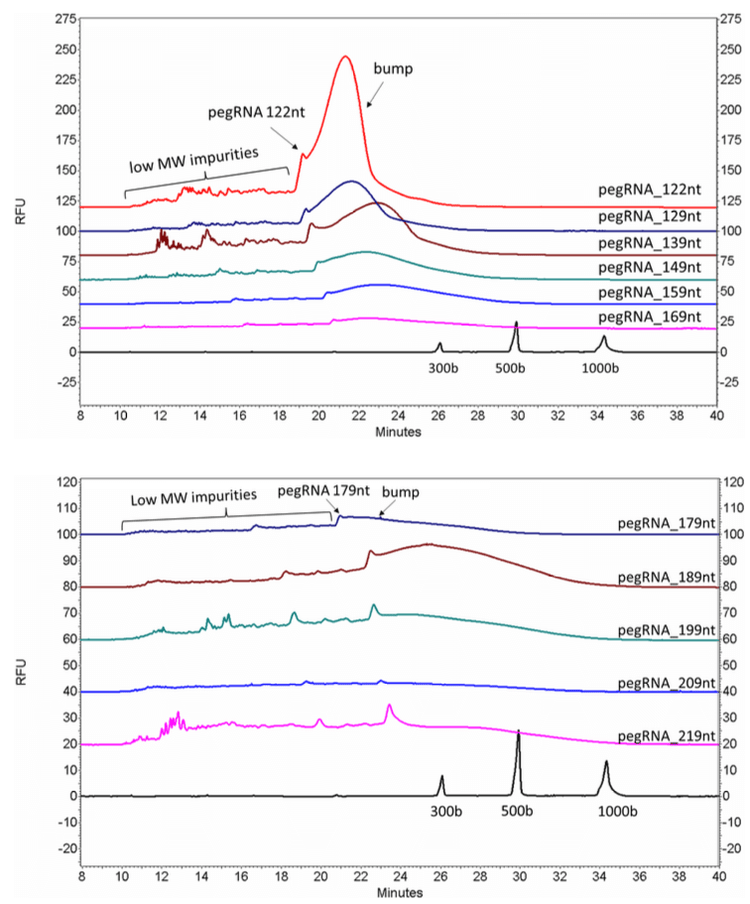
pegRNA oligonucleotides with lengths of 122 nt, 129 nt, 139 nt, 149 nt, 159 nt, 169 nt, 179 nt, 189 nt, 199 nt, 209 nt and 219 nt, together with an RNA ladder (50–1,000 bases), were analyzed with an optimized gel matrix and the ssDNA 100-R kit (Figure 11). All the pegRNA samples show a sharp peak followed by a big bump.
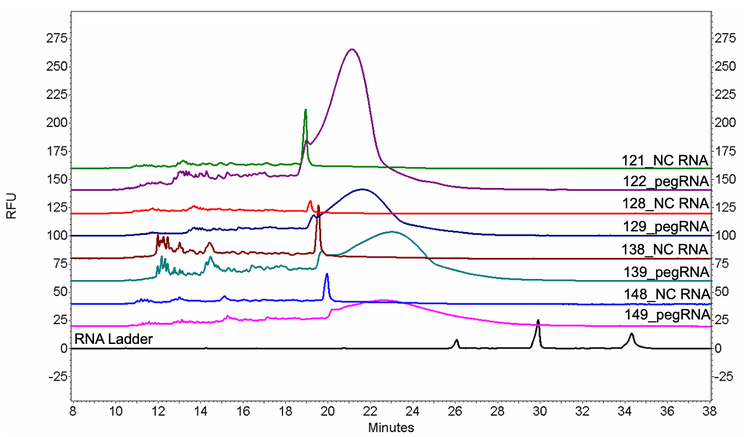
To investigate the root cause of the abnormal peak shape of pegRNAs, four non-complementary RNAs (NC RNAs) at various lengths (121 nt, 128 nt, 138 nt and 148 nt) were designed with the minimal possibility of forming secondary structures and analyzed
together with pegRNAs of similar lengths (122 nt, 129 nt, 139 nt and 149 nt). As shown in Figure 12, all NC RNAs showed a singular peak without the big bump, indicating that the abnormal peak shape of pegRNAs was probably due to their secondary structures.
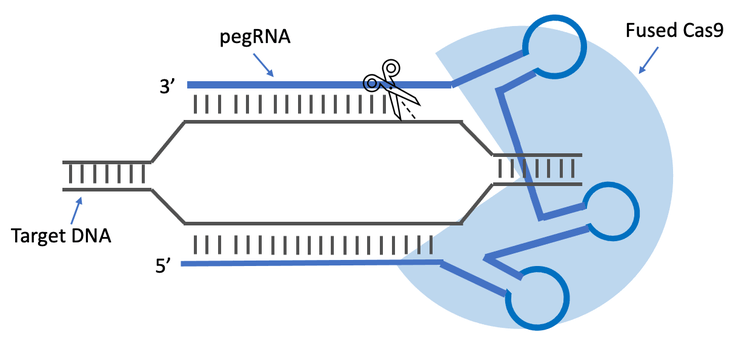
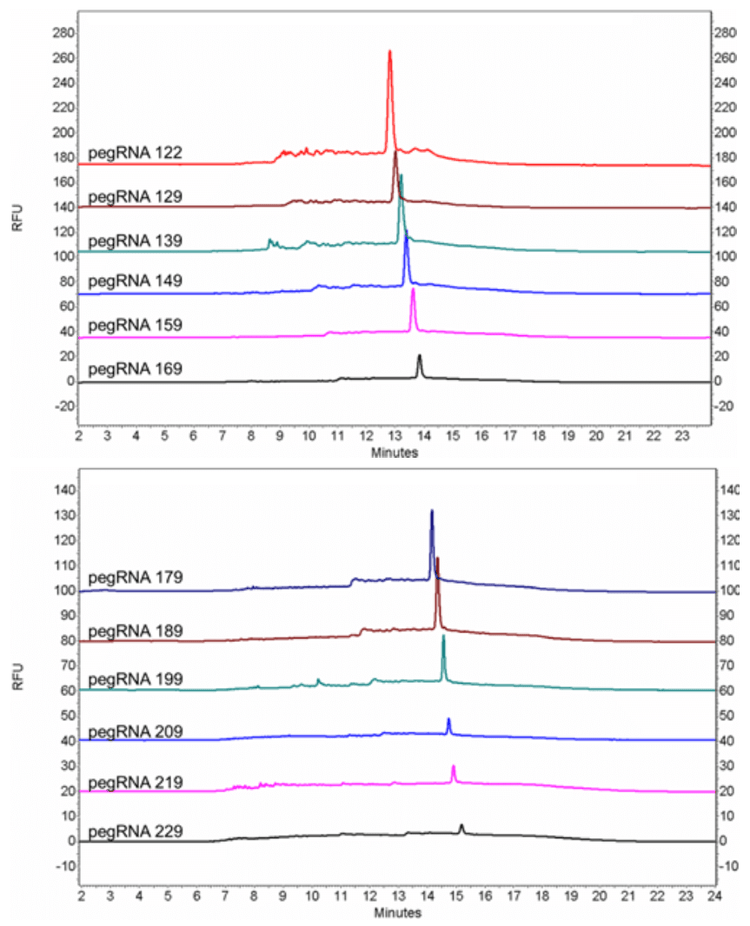
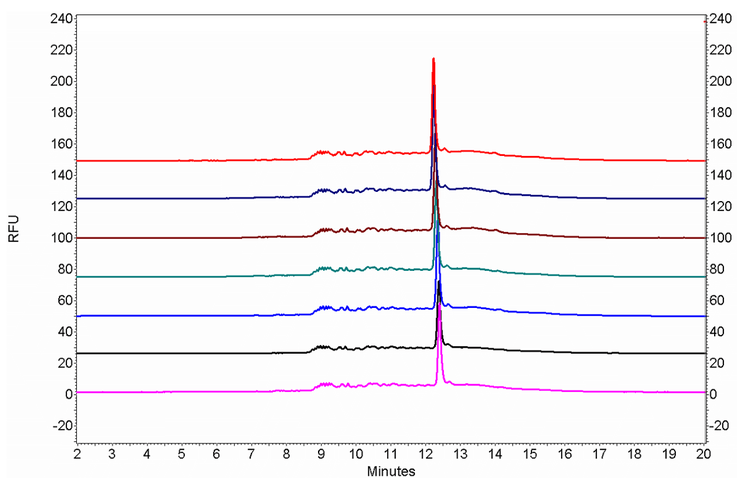
Therefore, the primary focus of this technical note was to develop a robust denaturing technique for pegRNA with high levels of secondary structure (Figure 13). After systematically exploring possible strategies for denaturing pegRNA, the optimized methods have been achieved by maintaining the denatured status of pegRNA during the entire separation process in addition to the one-step denaturing of pegRNA oligonucleotides. The pegRNA samples were prepared by mixing with formamide and heating at 65°C for 10 minutes. Then, the capillary temperature was increased to 50°C to maintain the pegRNA sample under a denatured state during the separation. As shown in Figure 14, a single, sharp peak has been obtained for all the tested pegRNA samples with the increased separation temperature.
A repeatability study of a representative pegRNA with a size of 129 nt was performed by seven consecutive injections. As shown in Figure 15, peak profile, migration time and signal level were very consistent between the seven injections. The RSD% of migration time (MT) and the corrected peak area% (CPA%) of FLP were 1.61% and 2.45%, respectively. The liquid cooling feature of the capillary temperature on the PA 800 Plus system enables stable and accurate temperature control during the separation, therefore providing a robust platform for maintaining the denatured status of pegRNA oligonucleotides. Accurate and repeatable purity analysis of pegRNAs was achieved after establishing the optimized sample treatment and capillary electrophoresis conditions.
Conclusion
An easy and fast workflow for the purity analysis of complex pegRNA molecules has been described in this technical note. High levels of sequence complementarity in pegRNAs result in remarkable overlapping higher-structure isoforms, presenting a considerable obstacle for purity analysis. After trialing various denaturation methods, the ability to resolve pegRNA sequences into a single, sharp peak has been achieved and %FLP can be determined. A stable and high temperature enabled by the capillary temperature control system on the PA 800 Plus system facilitates the maintenance of the denatured state of pegRNAs during the entire separation process, leading to optimal accuracy and repeatability with RSD% of <3% for the purity determination of pegRNA. It provides a convenient and robust method for quality assessment of chemically synthesized pegRNA within 30 minutes per sample.
References
- Anzalone, A.V.; Randolph, P.B.; Davis, J.R. et al. Search-and-replace genome editing without double- strand breaks or donor DNA. Nature 576, 149–157 (2019).
- Scholefield, J.; Harrison, P.T. Prime editing – an update on the field. Gene Ther. 28, 396–401 (2021).
- Matsoukas, I.G. Prime Editing: Genome Editing for Rare Genetic Diseases Without Double-Strand Breaks or Donor DNA. Front. Genet. 11:528 (2020).
- ssDNA 100-R kit. SCIEX application guide, RUO-IDV- 05-11137-A.