Abstract
This technical note demonstrates the power and capability of the SCIEX 7500+ system to sensitively detect and quantify lipids using a comprehensive lipidomics panel with a multiple reaction monitoring (MRM) scan mode. The panel consists of ~2000 lipid molecular species identified at the fatty acid level of structural specificity and depends on chromatography to separate lipids by class to mitigate isobaric interference. Due to lipids eluting as a class, the speed at which individual MRM transitions are measured is crucial to ensure good coverage and quantitative accuracy. A balance must be struck between the extent of the target list and the resulting data quality, especially during periods of high MRM concurrency. However, the new SCIEX 7500+ system with fast MRM technology addresses this problem. It is ideal for large panel quantitative screening to provide more data points across individual peaks to improve peak shapes and the calculated %CVs for quantitative measurements.
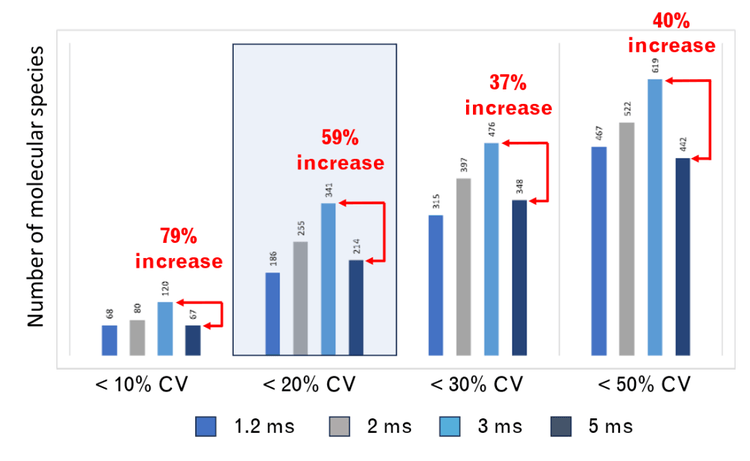
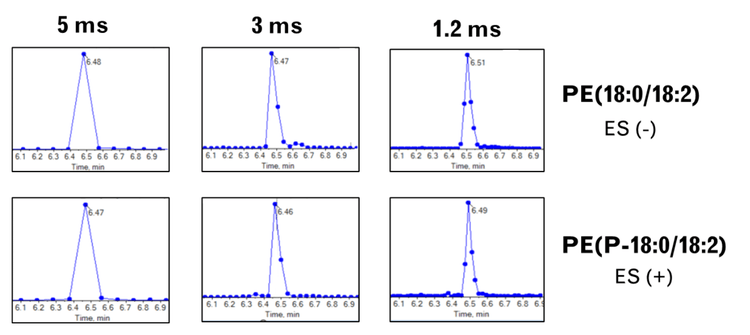
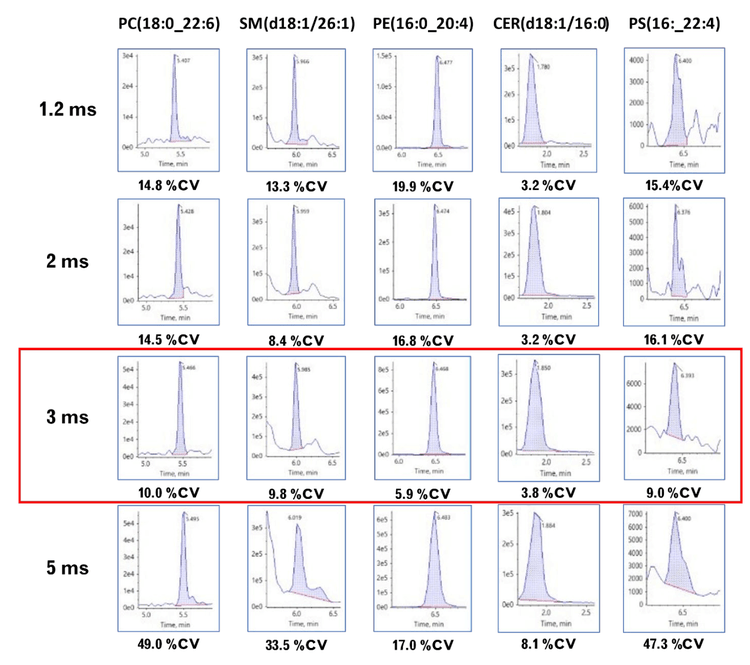
Here, the SCIEX 7500+ system was used to compare the quantitative performance of lipid analysis using different dwell and pause times. The fastest recommended speeds on the classic SCIEX 7500 system are a 2 ms dwell time and a 3 ms pause time (indicated here as a 5 ms scan speed). In contrast, the SCIEX 7500+ system can scan with settings as fast as 0.5 ms dwell and 0.7 ms pause (1.2 ms scan speed).Our findings reveal that using a combined scan time of 3 ms (1.5 ms dwell and 1.5 ms pause times) on the SCIEX 7500+ system resulted in data with superior quantitative precision compared to the classic SCIEX 7500 system. This advantage relative to the classic system was most evident during periods of high MRM concurrency (Figure 1). The faster data acquisition led to more data points across analyte peaks, which generated better peak shapes and improved lipid isomer resolution compared to the SCIEX 7500 system. These results demonstrate that the new instrument is capable of extremely fast analysis times and generates data of superior quality for lipid analysis.
Key features of lipidomics analysis using the SCIEX 7500+ system
- The ~4X faster analytical speed of the SCIEX 7500+ system improves the quantitative performance of a large, broadly targeted lipid panel
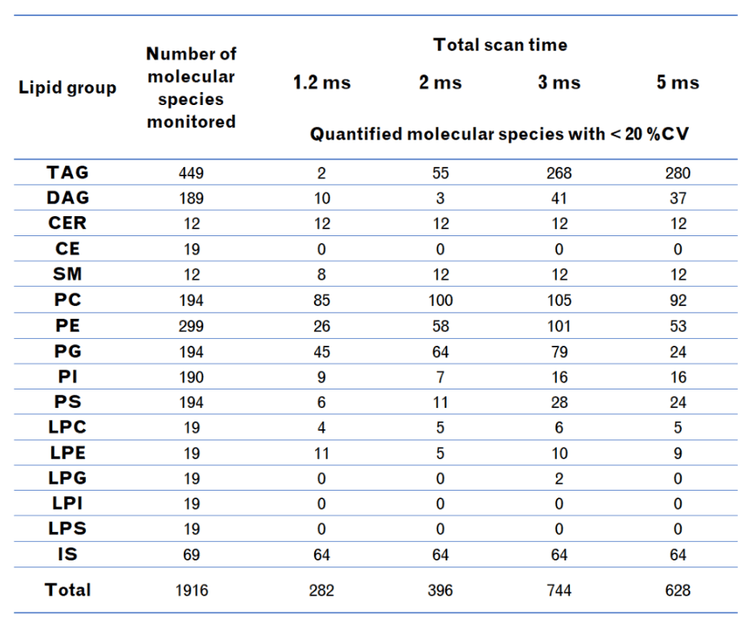
- The number of lipid molecular species quantified with %CV <20% increased 59% during a period of the highest MRM concurrency (n = 925) compared to the fastest speed recommended on the SCIEX 7500 system
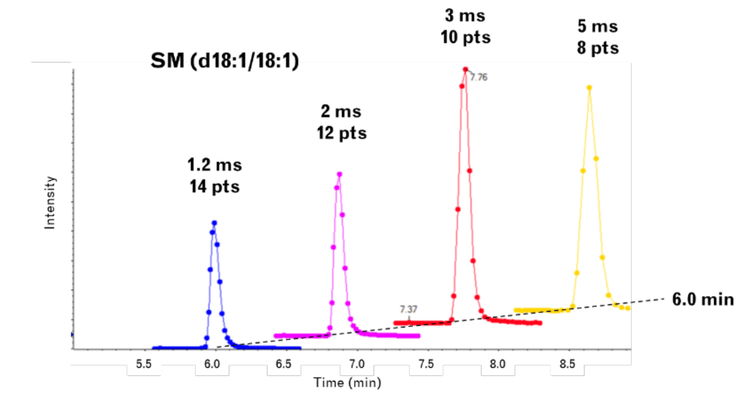
- The faster MRM acquisition rates improve peak shapes by increasing the number of data points across the peak and providing better resolution of near-eluting lipid isomers
Introduction
The analysis of lipids is a challenging analytical process. Although there are believed to be >150,000 individual lipid molecular species in the biome, a targeted or untargeted experiment typically only detects a small fraction in a biological sample. This result can be attributed to insufficient primary reference and internal standards, large lipid isomers and isobars, and limited information derived from collision-induced dissociation (CID). The quantitative analysis of lipids is further complicated by high signal variance, chromatographic challenges associated with separating a chemically diverse population of molecules and a broad (10–12 orders of magnitude) linear dynamic range1.
Mass spectrometry is the primary tool used for several lipid analysis approaches. The traditional shotgun lipidomics approach is an infusion method that identifies lipids based on common fragment ions or neutral losses from precursor ions generated as molecules undergo CID-based fragmentation2. This method generates data at the sum composition level (class identification in addition to a sum of the carbons and double bonds within the acyl chains of the lipid). This data type lacks specificity and has generally been replaced with targeted lipid analysis by HPLC ESI-MS/MS or untargeted lipid analysis, such as data-dependent acquisition (DDA). The latter method relies on software to interpret the data, and the results are not quantitative3. However, with an appropriate method, a targeted analysis can generate quantitative lipid data from diverse sample types4.
A comprehensive targeted lipid panel has been developed that bridges the gap between DDA discovery experiments and the need for accurate quantitation5,6. This method broadly targets >2250 lipid molecular species among 21 different lipid classes identified at the fatty acid level of structural specificity. This method determines the lipid class and associated fatty acids but cannot be used to determine the stereochemistry or location of double bonds. HILIC-like chromatography provides class separation to mitigate inter-lipid class isobaric overlap, and curated MRM transitions provide specific identification without the need for software designed to interpret DDA lipidomics data. This approach is limited by its dependence on a triple quadrupole mass spectrometer (TQMS) that is fast, sensitive and capable of rapid polarity switching. These features are required to accommodate the increasing number of targeted lipids in this broadly based quantitative screening experiment.
Earlier versions of high-end TQMS systems from SCIEX, namely the SCIEX Triple Quad 6500+ system and SCIEX 7500 system, have proven capable of the demands of this assay. The improved sensitivity of the SCIEX 7500 system increased the lipid coverage compared to the SCIEX Triple Quad 6500+ system. However, with both instruments, a balance was needed between the number of MRM transitions and the desired quantitative accuracy of their measurement. The 2 most significant variables to determine the time required for the instrument to scan through the entire list of MRMs (duty cycle) are dwell time and pause time. Dwell time is the time set for counting ions that hit the detector at a particular m/z. The longer the dwell time, the better the analyte peak's signal-to-noise (S/N) ratio. Pause time refers to the time needed to clear the ion path of precursor and fragment ions associated with the previous MRM transition and the time required for an ion of a particular m/z to pass through to the detector. Because the detector records ions and does not identify them, the ion path must be clear of residual ions from the previous scan to ensure accurate identification and quantitation, reducing signal enhancement from crosstalk. The classic SCIEX 7500 system has a top recommended scan speed of 2 ms dwell and 3 ms pause per MRM transition. For this technical note, these values are summed to give an aggregate dwell/pause time of 5 ms. In this assay, the average peak width, except for the TAGs and DAGs, is ~10 s, so the cycle time is limited to approximately 1 s. Most analysts opt for fewer points across the peak in lieu of better coverage, resulting in reduced quantitative accuracy. The SCIEX 7500+ system has improved ion transfer rates related to the octupole fields within Q0, accelerating ions and enabling faster ion transit times than previous ion optic configurations. As a result, the instrument is capable of unprecedented scan rates, achieving scan speeds as fast as 0.5 ms dwell and 0.7 ms pause (1.2 ms) without crosstalk between MRM transitions.
Here, the speed of the SCIEX 7500+ system was leveraged to improve the quantitative precision of a large lipid panel containing ~2000 lipids. The faster speed significantly improved the variance of the quantitative results, which increased the number of lipid molecular species identified and quantified with a %CV <20% by almost 60% (Figure 1). These improvements result from better peak shape due to more data points across the analyte peak. Overall, the large lipid panel analysis was improved significantly at all speeds faster than those recommended using the classic SCIEX 7500 system, demonstrating that the SCIEX 7500+ system is a significant advancement in the technology used to study lipids.
Methods
Materials: UltimateSPLASH ONE (Avanti Polar Lipids, Alabaster, AL) was used for internal standards. This mixture contains 69 deuterated standards and was specifically designed for this lipid panel method. By using multiple internal standards per lipid class, the differential fragmentation efficiency of lipids due to the number of carbons and double bonds in the acyl chains can be accommodated, which enables an internal strategy termed “accurate quantitation.” This internal strategy has recently been validated4. Bovine total heart lipid extract (Avanti Polar Lipids) was used as the 3 sample. This lipid extract contains all the major lipid classes; additionally, it is rich in odd-chain fatty acids, polyunsaturated fatty acids and plasmalogens, which provide a rich substrate for analysis. All solvents used were LC-MS grade and obtained from Honeywell-Burdick and Jackson (Muskegon, MI). Ammonium acetate was obtained from Sigma Aldrich (St. Louis, MO) and was prepared as a 2M stock solution in methanol. It is highly recommended that methanol from Burdick and Jackson be used, as many other solvent brands have significant chemical background contamination.
Sample preparation: Total bovine heart extract was diluted in 50:50 (v/v), methanol/dichloromethane with a serial dilution using a 10 µg/mL concentration stick solution. The resulting solution was then diluted 1:100 with mobile phase A to a final concentration of 1 µg/mL of lipid extract. An aliquot of internal standard was added to each sample to achieve a final dilution of 1:1000. Note that the prepared samples contained 2mM ammonium acetate, which can catalyze sample degradation in samples in approximately 3–4 days. Freshly prepared samples that contain ammonium acetate are essential.
Chromatography: Lipid mixtures were resolved using a Shimadzu Nexera Prominence HPLC system equipped with a Phenomenex Luna™ NH2 column (100 x 2.1 mm, 2.6 µm particle size). The autosampler sample bay was maintained at 15°C, and a 5 µL sample volume was injected on column. The column oven temperature was kept at 40°C with a constant mobile phase flow rate ranging from 0.2 to 0.7 mL/min for a total run time of 17 min (Table 1). Mobile phase A was 7:93 (v/v), dichloromethane/acetonitrile containing 2mM ammonium acetate. Mobile phase B was 50:50 (v/v), water/acetonitrile containing 2mM ammonium acetate. Note that the pH of mobile phases A and B was not adjusted. The chromatographic gradient details are shown in Table 1.



Results and discussion
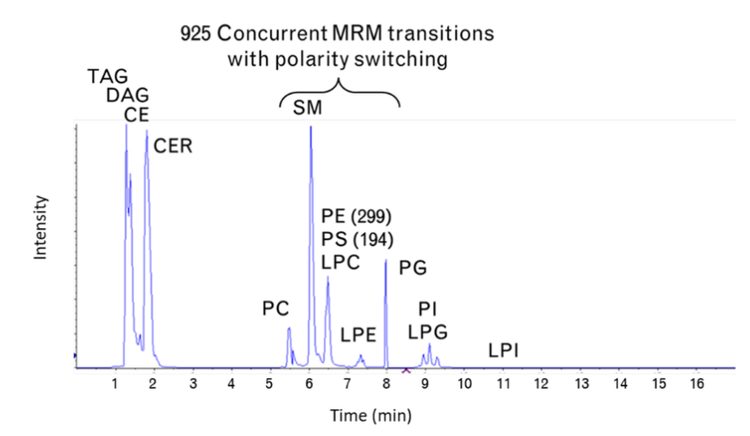
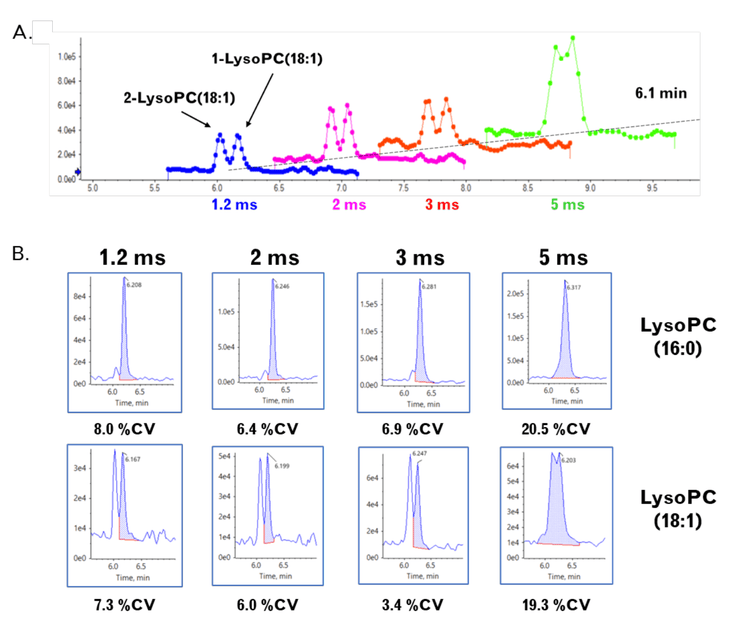
A key aspect of the large lipid panel method is the clear resolution of lipid classes by chromatography. This is important because the isobaric and isomeric overlap between lipid classes is common in lipid analysis. Many lipids from different classes have similar or the same mass; those same molecules can share common fragments or neutral losses. These similarities make the interpretation of DDA data by software difficult. This challenge can be mitigated by separating the lipid classes and, therefore, enabling an MRM transition to detect and differentiate lipid molecular species from isomers. For example, the isomers PC(14:0_20:1) and PC(16:0_18:1) can be selectively detected using MRM transitions that target the respective fatty acids with the Q3 transition.
Figure 2 shows the total ion chromatogram (TIC) that results from using the NH2 column. Like true HILIC chromatography, this column separates the different lipid classes. Unlike other HILIC columns, the NH2 column is hydrophobic enough to separate the neutral lipid classes and improve specificity. Using the NH2 column resulted in the highest MRM concurrency between 5.5 and 8 minutes. During this time, 925 MRM transitions were monitored. The effects of fast-scanning MRM on peak shape and quantitative precision were expected to be most evident in this region of the chromatogram.
Conclusion
- The SCIEX 7500+ system is capable of measuring analytes at a combined speed of 1.2 ms per MRM transition, enabling an 800 MRM/s scan speed
- Faster data acquisition enables the collection of more data points across an analyte peak, which improves the analyte peak shape and the quantitative accuracy of the experiment when integrated
- The increased data point coverage across analyte peaks and the improved peak shape can improve the resolution of closely eluting isomers
- In this extensive panel of lipids, using a combined scan speed of 3 ms (1.5 ms dwell, 1.5 ms pause) produces more accurate quantitative data compared to data obtained at 5 ms, which is the fastest recommended scan speed on the classic SCIEX 7500 system
- There is a 60% increase in the number of molecular species measured with a %CV <20% using the SCIEX 7500+ system at a scan speed of 3 ms compared to that achieved using a 5 ms scan speed during the period of highest MRM concurrency
References
- Heiles S. Advanced tandem mass spectrometry in metabolomics and lipidomics-methods and applications. Anal Bioanal Chem. 2021 Oct;413(24):5927-5948. doi: 10.1007/s00216-021-03425-1. Epub 2021 Jun 18. PMID: 34142202; PMCID: PMC8440309.
- Han X, Gross RW. The foundations and development of lipidomics. J Lipid Res. 2022 Feb;63(2):100164. doi: 10.1016/j.jlr.2021.100164. Epub 2021 Dec 22. PMID: 34953866; PMCID: PMC8953652.
- Cajka T, Fiehn O. Toward Merging Untargeted and Targeted Methods in Mass Spectrometry-Based Metabolomics and Lipidomics. Anal Chem. 2016 Jan 5;88(1):524-45. doi: 10.1021/acs.analchem.5b04491. Epub 2015 Dec 16. PMID: 26637011.
- Zhang Z, Singh M, Kindt A, Wegrzyn AB, Pearson MJ, Ali A, Harms AC, Baker PRS, Hankemeier T. Development of a targeted hydrophilic interaction liquid chromatographytandem mass spectrometry based lipidomics platform applied to a coronavirus disease severity study. J Chromatogr A. 2023 Oct 11;1708:464342. doi: 10.1016/j.chroma.2023.464342. Epub 2023 Aug 31. PMID: 37696124.
- Achieve broad lipid quantitation using a high-throughput targeted lipidomics method. SCIEX technical note, RUOMKT-02-8482-B.
- Comprehensive Targeted Method for Global Lipidomics Analysis. SCIEX technical note, RUO-MKT-02-8477-B.
- Tan ST, Ramesh T, Toh XR, Nguyen LN. Emerging roles of lysophospholipids in health and disease. Prog Lipid Res. 2020 Nov;80:101068. doi: 10.1016/j.plipres.2020.101068. Epub 2020 Oct 15. PMID: 33068601.