Abstract
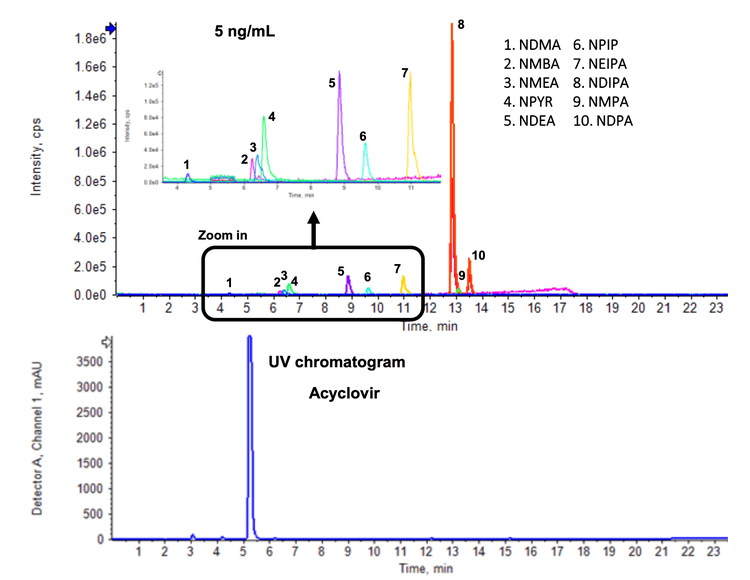
This technical note demonstrates a highly selective and sensitive method for quantifying 10 nitrosamines in acyclovir. Baseline chromatographic separation was achieved between the 10 nitrosamines and the acyclovir active pharmaceutical ingredient (API) (Figure 1). Low-level quantification was achieved in spiked API samples. Accurate and highly reproducible quantitative performance was reached, meeting critical requirements for nitrosamine analysis using the QTRAP 6500+ system (Figure 2). Linearity was reached across a wide linear range covering 0.025-20 ng/mL.

Introduction
Nitrosamines are a class of probable carcinogenic and mutagenic compounds that have caused the recall of more than 100 compounds since 2018.1 This includes the recent recall of acyclovir, a pharmaceutical product used to treat viral infections such as shingles and chickenpox. The US FDA has amended the maximum permissible daily intake limits for nitrosamine compounds, which for most drug products is now 30 ng/g. This is equivalent to a maximum daily dose of less than 880 mg/day.2-3 Given that acyclovir has a maximum daily dose of 800 mg, a nitrosamine limit of 30 ng/g can be implemented.
Key features of nitrosamine analysis using the QTRAP 6500+ system
- Sensitive quantification: Achieve low-pg/mL level quantification of 10 critical nitrosamine impurities in acyclovir API using minimal sample preparation
- Baseline chromatographic separation: Reach accurate quantification with baseline separation of 10 nitrosamines and acyclovir API in a single injection
- Effortlessly meet important quantitative performance criteria: Achieve accurate and highly reproducible quantification of nitrosamines with strong linearity using the QTRAP 6500+ system
- Streamlined data management: Data acquisition and processing are simplified with SCIEX OS software, a 21 CFR Part 11-compliant platform
Methods
Standard preparation: Each nitrosamine was mixed at 100 µg/mL and diluted in water to generate a 1000 ng/mL mixture.
Sample preparation: A 100 mg (±5 mg) sample of the acyclovir API was weighed into a suitable vessel. A 2.5 mL aliquot of water was added, shaken to mix and then vortexed for 30 seconds. The solution was sonicated for 15 minutes and centrifuged at 3901 RCF for 5 minutes. The supernatant was removed and filtered through a 0.2 µm PTFE filter and transferred to an HPLC vial for analysis. The resulting sample concentration was 40 mg/mL.
Spiked sample preparation: A 100 mg (±5 mg) sample of the API was weighed into a suitable vessel. A 2.5 mL aliquot of a 1 ng/mL nitrosamine mix standard solution was added before being shaken by hand. The sample was vortexed for 30 seconds and centrifuged at 4500 rpm for 5 minutes. The supernatant was removed and filtered through a 0.2 µm PTFE filter and transferred to an HPLC vial for analysis. The final concentration was 40 mg/mL with a spike concentration of 1 ng/mL of the nitrosamine mix. Each nitrosamine was present at a spike concentration of 25 ng/g relative to the sample.

Chromatography: An ExionLC system with a Phenomenex Luna Omega Polar C18 column (3.0 x 150 mm, 3.0 µm, 100 Å) was used for separation at a flow rate of 0.45 mL/min. The operating column temperature was 35°C. Mobile phase A was 0.1% formic acid in water and mobile phase B was 0.1% formic acid in methanol. Table 1 summarizes the gradient conditions used. The injection volume was 20 µL.
Mass spectrometry: The QTRAP 6500+ system was operated in positive ion mode using APCI ionization. The data was collected using an MRM method. Table 2 outlines the source and gas parameters used. The compound-specific MRM parameters used are shown in Table 3.


Quantitative performance
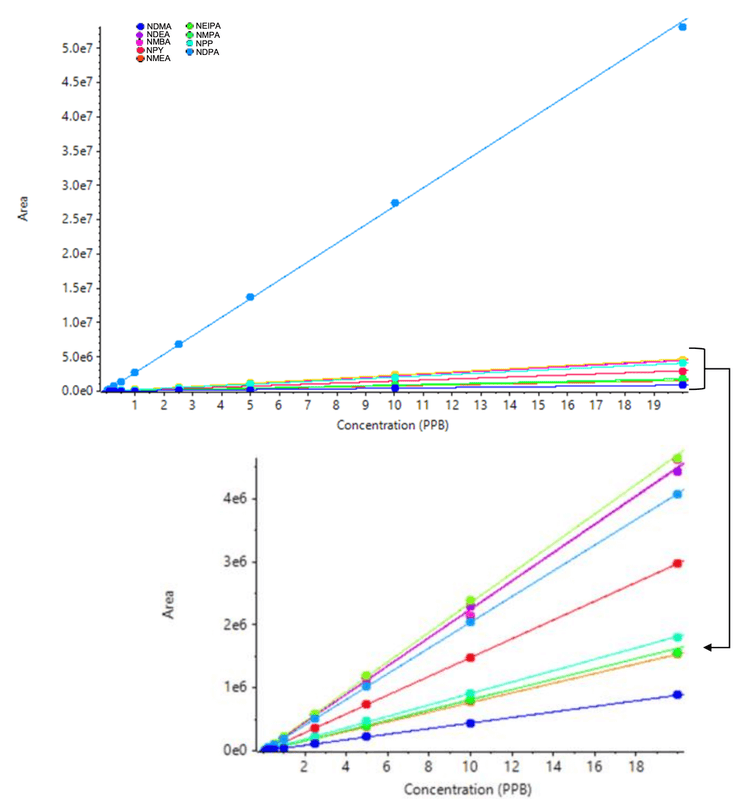
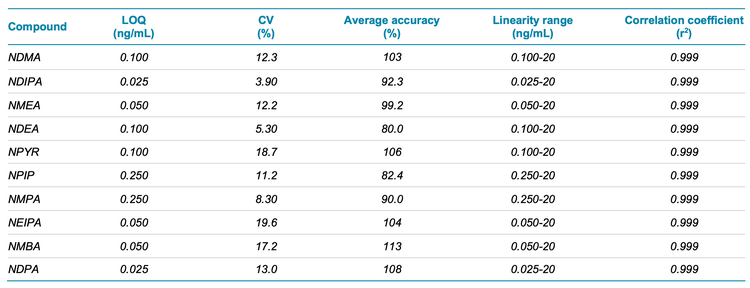
The calibration curves for 10 nitrosamines were plotted across a concentration range of 0.025-20 ng/mL (Figure 2). Strong linearity was achieved with correlation coefficients >0.99 for all compounds analyzed (Table 4). The linear dynamic range was greater than 3 orders of magnitude for all nitrosamines.
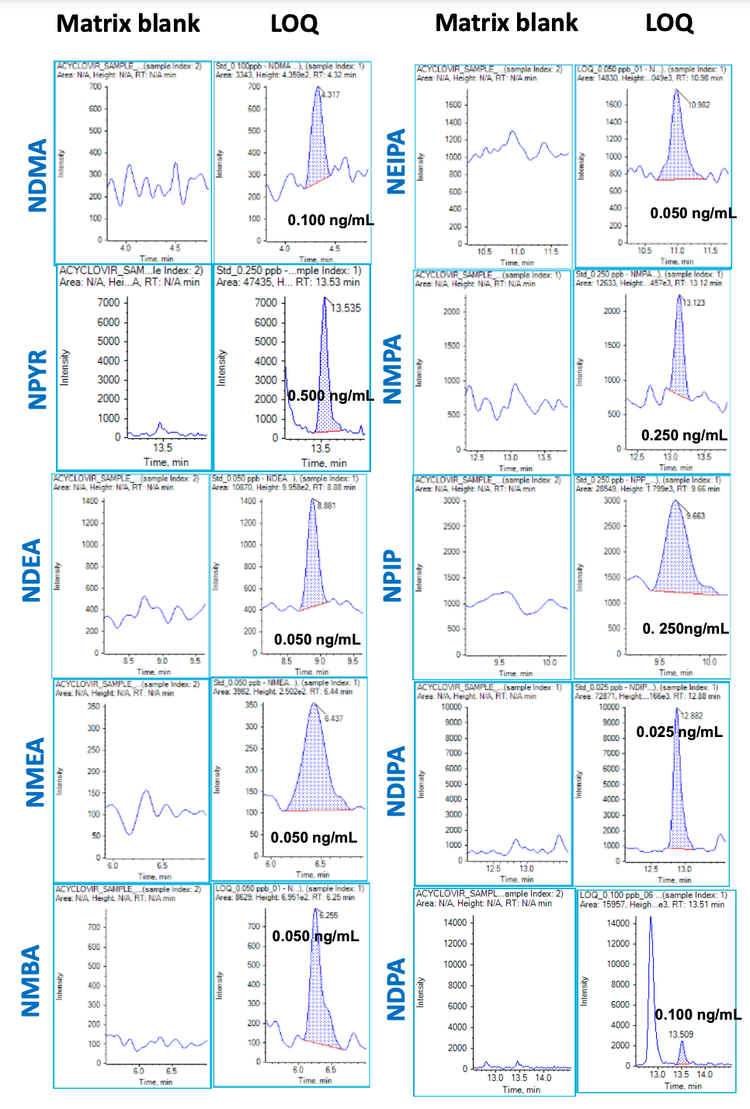
Each LOQ level was evaluated in 6 replicates. The XICs at the LOQ and the matrix blank for all 10 nitrosamines are highlighted in Figure 3. Interferences were not observed in the matrix blanks for any of the nitrosamines analyzed (Figure 3). The observed accuracy and precision levels met the specified regulatory recommendations for nitrosamine analysis. The overall coefficient of variation (CV) was <20% and accuracy was within ±20% of the nominal concentration at the LOQ level. The quantitative performance for each nitrosamine is summarized in Table 4.
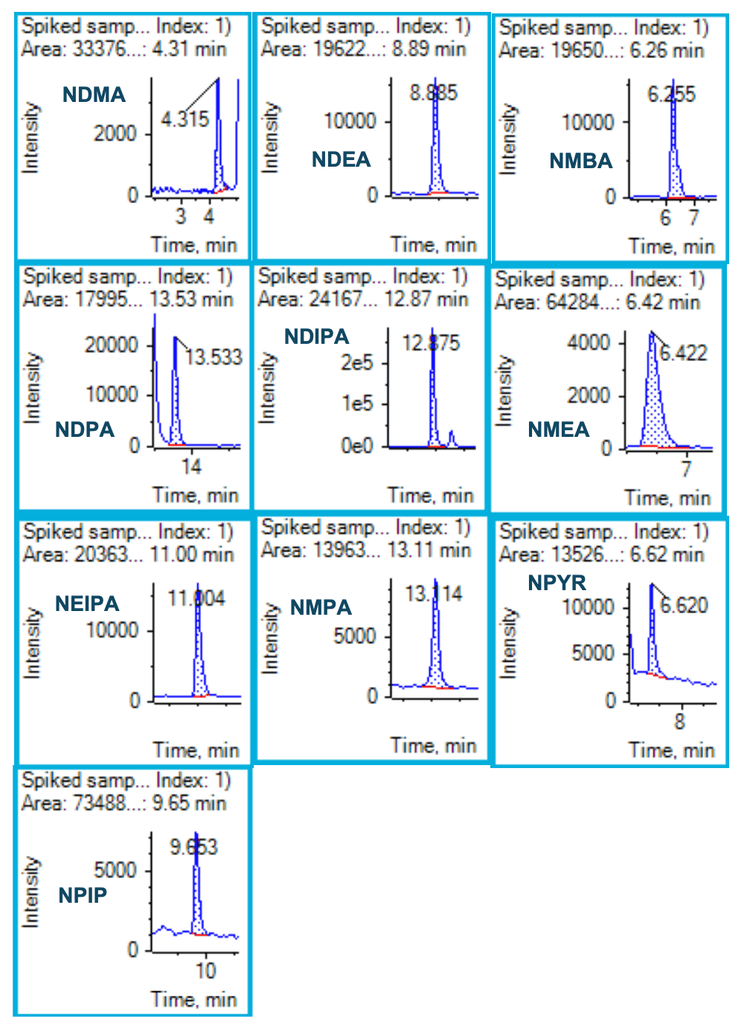
Quantification of spiked concentrations in samples
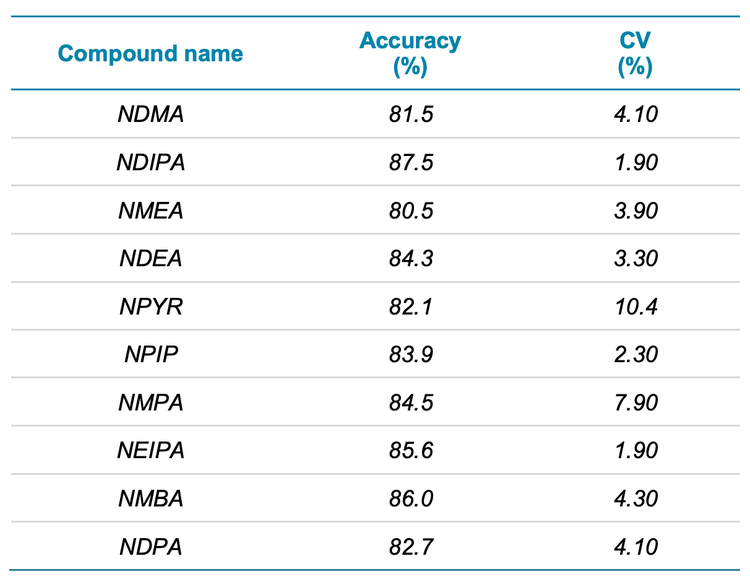
Accuracy and precision metrics were evaluated in standard solutions and spiked samples. A 1 ng/mL concentration in spiked solution (equivalent to 25 ng/g in sample concentration) was used for the assessment. Figure 4 shows the representative XICs of the spiked sample equivalent to a 25 ng/g concentration. The acceptable criteria for accuracy and precision at this concentration level are ±30% of the nominal concentration and ≤25% for %CV. The overall accuracy was within ±20% of the nominal concentration and the %CV was <20%, meeting the specified requirements. Table 5 summarizes the accuracy and %CV for each nitrosamine in spiked samples. The spike concentration was below the recommended limit (30 ng/g), indicating that the assay meets the specified requirements for nitrosamine analysis. Accuracy was within ±19.5% of the nominal concentration with %CV <10.4%.
Compliant-ready SCIEX OS software
To meet the regulations outlined in 21 CFR Part 11, SCIEX OS software is a closed system and requires records and signatures to be stored electronically. SCIEX OS software can open raw data files from any visible storage location within a closed network by using designated processing workstations. Figure 5 illustrates 3 types of controls that are required for 21 CFR Part 11 compliance. The workflow presented here is fully compliant with these guidelines, as SCIEX provides 1) technical controls over hardware and software configuration, 2) network security and secure operating systems and policies and 3) procedures and user training (Figure 5).

Conclusion
- Low-level quantification was achieved for 10 nitrosamines in spiked samples using the QTRAP 6500+ system
- Good chromatographic separation was achieved between the 10 nitrosamines and the acyclovir API
- Accurate and highly reproducible quantitative performance was demonstrated for all 10 nitrosamines with strong linearity, meeting the specified acceptance criteria
- The method demonstrated the quantification of nitrosamine impurities below the current recommended limit (30 ng/g) in the acyclovir drug product
- Using a straightforward sample preparation, multiple preparations of the un-spiked sample generated consistent results • A single platform for streamlined data acquisition, processing and management with SCIEX OS software was presented
References
- Control of Nitrosamine Impurities in Human Drugs, February 2021, US food and drug administration Guidance for Industry, Revision 1
- Nitrosamine impurities in medications: Recalls, Government of Canada
- Update on nitrosamine impurities: EMA continues to work to prevent impurities in medicines, EMA/241020/2019, 26th April 2019