Abstract
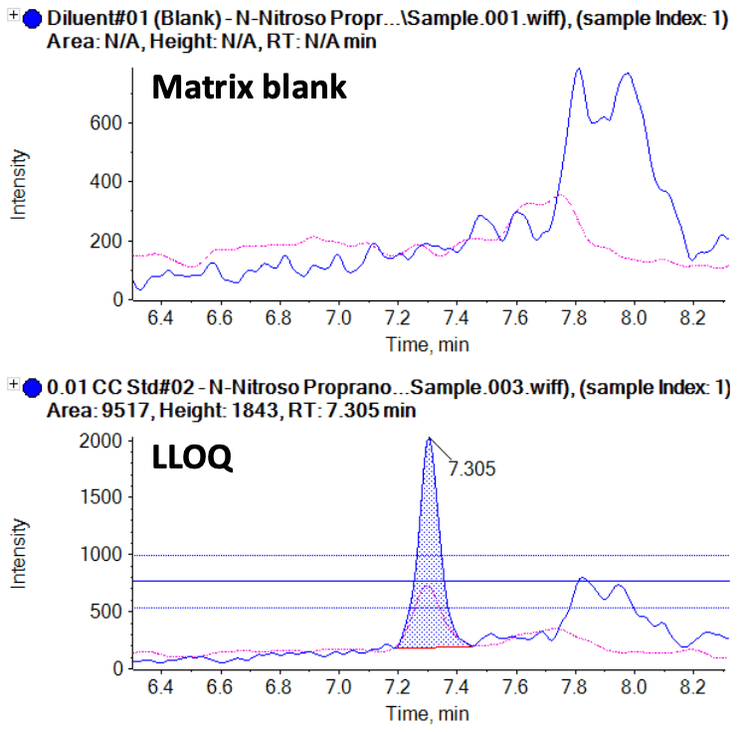
This technical note describes the quantification of the N-nitroso propranolol impurity in a propranolol drug substance and product using the QTRAP 6500+ system. A lower limit of quantification (LLOQ) of 0.01 ng/mL was achieved for Nnitroso propanol with high reproducibility and accuracy (Figure 1). In addition, linearity was achieved between 0.01 ng/mL and 10.00 ng/mL, providing a linear dynamic range (LDR) of 3 orders of magnitude.
Introduction
Propranolol, a synthetic amino alcohol, is a competitive nonselective, β-adrenoreceptor antagonist extensively used to treat hypertension, angina pectoris and other cardiac diseases. 1 β-adrenergic blocking drugs such as propranolol, atenolol, metoprolol, nadolol, oxprenolol and sotalol contain amine groups. As a result, these drugs react with sodium nitrite in a hydrochloric acid solution to produce N-nitrosamines. 2 In March 2022, N-nitroso propranolol was reported in various strengths of the propranolol hydrochloride extended-release capsule. Long-term exposure to N-nitroso propranolol at unsafe levels might increase cancer risk. 3
Key features for the quantification of the Nnitroso propranolol impurity
- Sensitive and selective quantification: Accomplish lowlevel quantification of the N-nitroso propranolol impurity using the QTRAP 6500+ system
- Effortlessly meet critical quantitative performance criteria: Achieve accurate and highly reproducible quantification of N-nitroso propanol using the QTRAP 6500+ syste
- High extraction recovery: Reach good recovery for Nnitroso propranolol in both the drug substance and drug product using a simple sample preparation workflow
- Streamlined data management: Data management and processing are simplified with SCIEX OS software, a 21 CFR Part 11-compliant platform
Methods
Standard preparation: A 1 mg/mL N-nitroso propranolol solution was prepared in methanol. The stock solution was diluted with 80:20 (v/v), acetonitrile/water to prepare calibration standards at concentrations ranging from 0.005 to 10 ng/mL that were stored under refrigerated conditions.
Drug substance: Propranolol was weighed and diluted to a final concentration of 1 mg/mL in 80:20 (v/v), acetonitrile/water. The sample was shaken for 20 minutes using a mechanical wrist action shaker. After extraction, the sample was centrifuged for 10 minutes at 4000 rpm. The supernatant was collected and filtered using a 0.22 μm PVDF syringe filter. The filtered solution was transferred into an HPLC vial for analysis.
Drug product: The drug product was crushed and weighed. A 1 mg/mL solution of the drug product with respect to the active pharmaceutical ingredient (API) was prepared using 80:20 acetonitrile/water. The solution was vortexed for 2 minutes and shaken for 30 minutes using a mechanical wrist action shaker. After extraction, the sample was centrifuged for 10 minutes at 4000 rpm. The supernatant was collected and filtered using a 0.22 μm PVDF syringe filter. The filtered solution was transferred into an HPLC vial for analysis.
Chromatography: An ExionLC system was used with a Phenomenex Kinetex biphenyl column (3.0 x 150 mm, 2.6 µm, 100 Å) for chromatographic separation at a flow rate of 0.4 mL/min. The operating column temperature was 40°C. Mobile phase A was 1mM ammonium formate with 0.1% (v/v) formic acid in water. Mobile phase B was 0.1% (v/v) formic acid in acetonitrile. Table 1 summarizes the gradient conditions used. An injection volume of 15 µL was used for analysis.



Results
ESI in positive polarity offered highly sensitive signal intensities for N-nitroso propranolol. Several chromatographic gradients and columns of various chemistries were evaluated (data not shown) to optimize the selectivity and specificity for quantifying the N-nitroso propranolol impurity in the presence of excipients, drug-related impurities and N-formyl propranolol impurities. The Phenomenex Kinetex Biphenyl LC column (3.0 x 150 mm, 2.6 µm, 100 Å) was selected for separation, as it provided high separation power.
Under the optimal LC-MS/MS conditions, the N-nitroso propranolol impurity eluted at a retention time of 7.30 minutes. Matrix interferences were not observed in the diluent or control samples, demonstrating the selectivity and specificity of the assay. A limit of detection (LOD) of 0.005 ng/mL and LLOQ of 0.010 ng/mL for the N-nitroso propranolol impurity were achieved. The representative XICs for the matrix blank and the LOD, LLOQ and specification levels of the sample are shown in Figure 2. The signal-to-noise (S/N) ratios observed at the LOD and LLOQ levels were >10 and >20, respectively.
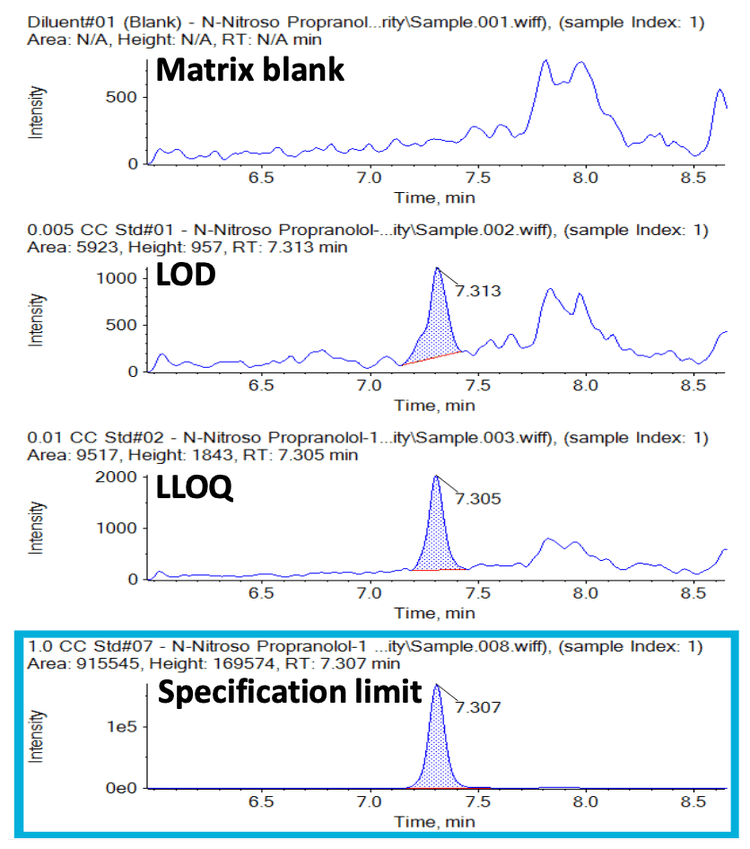
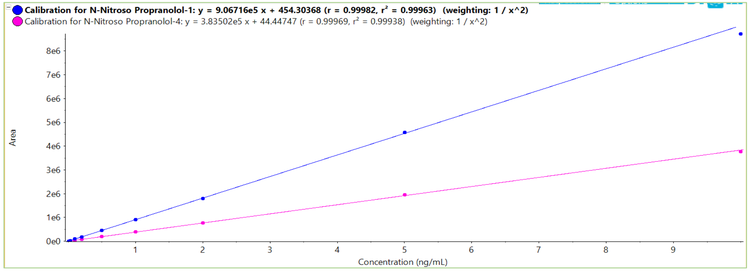
The upper limit of quantification (ULOQ) was selected based on the highest levels of analytes expected to be observed and is not a reflection of system capability. The calibration curve generated for the concentrations tested is shown in Figure 3 with an overlay of the linearities for both the quantifier and qualifier transition responses. Linearity was demonstrated over a concentration range of 0.01 ng/mL to 10.00 ng/mL. A correlation coefficient (r2) of >0.99 was achieved for responses from both transitions.
Accuracy was expressed as percentage deviation from the nominal value at each respective concentration level. The precision of the assay was measured by the percent coefficient of variation (%CV) at each of the concentrations. Accuracy and %CV were determined by analyzing 6 replicates of the samples at the LLOQ and specification limit in a single analytical run. As shown in Table 4, the %CV values at the LLOQ and specification limit were 6.74% and 3.59%, respectively. Accuracy ranged from 96% to 115% at the LLOQ and 107% to 116% at the specification limit, averaging 105% and 110%, respectively. Overall, these data confirm the accuracy and high reproducibility of the method for quantifying N-nitroso propranolol (Table 4).


Compliant-ready SCIEX OS software
SCIEX OS software is a closed system and requires records and signatures to be stored electronically, meeting the regulations outlined in 21 CFR Part 11. SCIEX OS software can open raw data files from any visible storage location within a closed network by using designated processing workstations. Figure 5 illustrates 3 types of controls required for 21 CFR Part 11 compliance. The workflow presented here is fully compliant with these guidelines, as SCIEX provides 1) technical controls over hardware and software configuration, 2) network security and secure operating systems and policies and 3) procedures and user training (Figure 5).

Conclusion
- Low-level quantification of the N-nitroso propanol impurity was achieved using the QTRAP 6500+ system
- Accurate and highly reproducible quantification of Nnitroso propanol was demonstrated. Linearity was achieved between 0.01 to 10 ng/mL, generating an LDR of 3 orders of magnitude
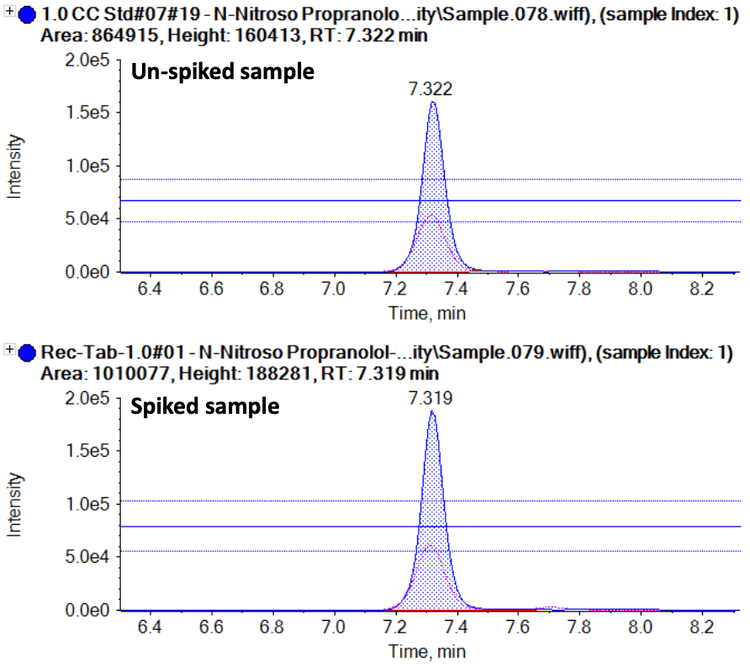
- The method demonstrated a simple sample preparation workflow with high recovery. The recovery was evaluated in placebo, API and drug product matrices.
- A single platform for streamlined data processing and management with SCIEX OS software was presented. The SCIEX OS software is also a 21 CFR Part 11- compliant platform that can be easily integrated into a regulated environment.
References
- Partani P. et al., Simultaneous determination of propranolol and 4-hydroxy propranolol in human plasma by solid phase extraction and liquid chromatography/electrospray tandem mass spectrometry. J Pharm Biomed Anal. 2009 Dec 5;50(5):966-76.
- Robbiano L, Martelli A, Allavena A, Mazzei M, Gazzaniga GM, Brambilla G. Formation of the N-nitroso derivatives of six beta-adrenergic-blocking agents and their genotoxic effects in rat and human hepatocytes. Cancer Res. 1991 May 1;51(9):2273-9.
- Recalls propranolol hydrochloride capsules due to a nitrosamine impurity - Canada.ca
https://recalls-rappels.canada.ca/en/alert-recall/pfizerrecalls-inderal-propranolol-hydrochloride-capsules-duenitrosamine-impurity - Control of nitrosamine impurities in human drugs – guidance for industry, FDA, February 2021.
- International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use; “ICH Harmonised Guideline - Assessment And Control Of DNA Reactive (Mutagenic) Impurities in Pharmaceuticals to Limit Potential Carcinogenic Risk, M7(R1)”; March 31, 2017
- Brambilla G, Martelli A, Sottofattori E. Nitrosation of propranolol under simulated gastric conditions. Carcinogenesis. 1995 May;16(5):1239-42.