Abstract
This technical note describes high-throughput quantitative proteomics using the SCIEX ZenoTOF 8600 system. Combining improved separations with the Evosep One system and high-sensitivity data-independent acquisition (DIA) of the ZenoTOF 8600 system, powered by ZT Scan 2.0 DIA, as many as 26,000 precursors and 4,000 protein groups were identified from 5 ng commercial human digest at 500 samples-per-day (SPD) throughput. This represents >5-fold improvement of precursor identifications and >3-fold improvement of protein group identifications at similar loads on previous-generation instrumentation, enabling a greater depth of proteome coverage for large-scale cohort analysis.
Key features of high-throughput quantitative proteomics on the ZenoTOF 8600 system
-
Sensitivity improvements on the ZenoTOF 8600 system result in LC-MS peak area gains as much as 10-fold relative to previous-generation instrumentation
-
ZT Scan 2.0 DIA enables gains in quantitative identifications for precursors and protein groups of >5-fold and >3-fold, respectively
-
Higher efficiency ion transmission and advanced optical detection on the ZenoTOF 8600 system result in more protein groups and precursors identified and quantified from 5 ng sample loadings compared to 200 ng loadings on previous generation QTOF instrumentation
-
Novel LC-MS methods enable unparalleled performance for quantitative proteomics at up to 500 SPD
Introduction
With the significant applications of high-throughput proteomics in translational and clinical research, it is important to consider the sensitivity and quantitative accuracy of these workflows. Quantitative proteomics is crucial because it allows researchers to move beyond merely identifying proteins to understanding their abundance and expression, which is essential for comprehending biological processes and identifying disease markers. Clinical studies must be conducted at scale and with precise measurement methods to achieve statistically meaningful results. Next-generation proteomics research has therefore shifted toward designing workflows with the ability to process large numbers of samples.
The SCIEX ZenoTOF 8600 system represents a significant leap forward in high-resolution mass spectrometry (HR-MS). A combination of the OptiFlow Pro ion source, higher-efficiency ion capture, and an advanced optical detector drives sensitivity improvements >10-fold over previous-generation HR-MS instrumentation. ZT Scan 2.0 DIA uses a continuously scanning quadrupole for precursor isolation, providing users with enhanced flexibility to refine methods for better qualitative and quantitative analysis compared to discrete-window DIA methods. When combined with improved separation techniques available on the Evosep One system, these advancements in the ZenoTOF 8600 system demonstrate exceptional performance for the quantitative identification of peptides and proteins in complex samples at throughputs of up to 500 SPD.
Methods
Sample preparation: Human K562 lysate tryptic digest standard was purchased from Promega and reconstituted in 0.1% formic acid in water. Samples (either 5 ng or 50 ng total amounts) were loaded onto Evotips as directed in the Evosep protocol.
Chromatography: Separations were carried out using an Evosep One system with optimized methods for 200, 300, and 500 SPD. An EV1182 column (4 cm x 150 µm, 1.9 µm particle size), heated to 40°C was used for all experiments. Samples were analyzed in triplicate for all loads and gradient lengths.
Mass spectrometry: Data-independent acquisition (DIA) analysis was carried out on a ZenoTOF 8600 system using an ion source configured with a 1-10 µL/min microflow electrode. The Evosep column was connected to the microflow probe using a PEEK 1/16” nanotight union (IDEX). Method parameters were as described in Table 1.
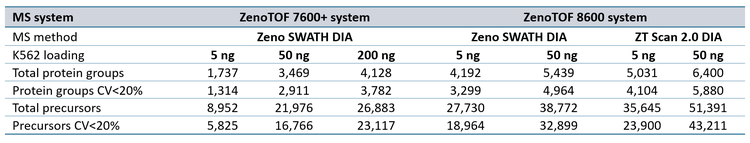
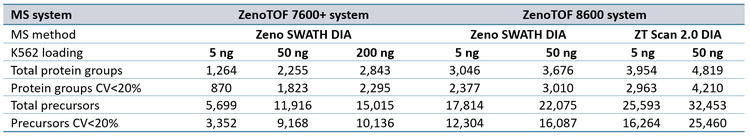
Data processing: DIA data were processed using either DIA-NN version 1.9.1 (1) or PEAKS Studio version 12.5 software to determine the number of protein groups and precursors. For processing with DIA-NN software, SCIEX data files (.wiff) were converted to .dia format using a research-grade data converter before processing. Database searches were performed using a K562/HeLa spectral library as previously described (2). Default parameter settings were used with the following exceptions: precursor m/z range was set to 400-900, fragment m/z range was set to 140-1750, and Match Between Runs (MBR) was used. Quantification strategy was set to “Legacy” mode. For ZT Scan DIA data processing, the “--scanning-swath” command option was used. Existing ZenoTOF 7600+ system data files for 5 ng, 50 ng, and 200 ng K562 loadings run at 200, 300, and 500 SPD (4) were processed as above for comparison against ZenoTOF 8600 system performance. Triplicate files for a given loading, SPD method, and DIA method were searched together. Protein groups and precursors were determined from the output “pg_matrix.tsv” and “pr_matrix.tsv” files generated by DIA-NN software.
For processing with PEAKS Studio software, a combination search was done using both the K562/HeLa library above as well as the human canonical FASTA protein sequences. Triplicate files for a given loading, SPD method, and DIA method were searched together. Mass ranges were set as above. Carbamidomethylation (C) was set as a fixed modification, while oxidation (M) and deamidation (N/Q) were set as variable modifications. Digest mode was set to semi-specific, with the number of missed cleavages set to 1. The number of protein groups and precursors was determined from the exported Protein CSV and Feature Vector CSV files, respectively.
Additional quantitation data processing was performed using the Analytics module of SCIEX OS software. Quantitation analysis was performed using fragment ions for 15 random peptides from human proteins identified in the K562 digest, choosing 2 fragment ions per peptide.

Gains in quantitative identifications of protein groups and precursors on the ZenoTOF 8600 system
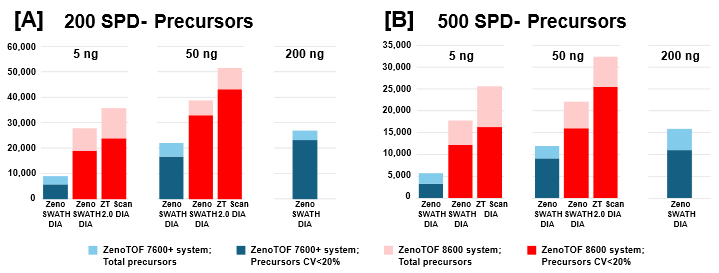
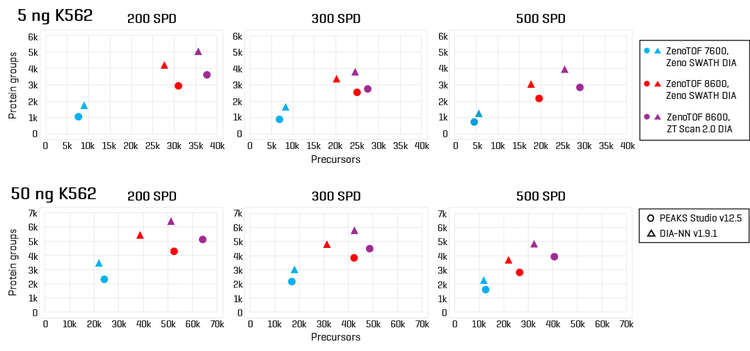
Sample loadings of 5 ng and 50 ng K562 digest were analyzed with 200, 300, or 500 SPD using the Evosep One and ZenoTOF 8600 systems. Zeno SWATH DIA methods employed 60 variable-width windows, with MS/MS accumulation times of 7 milliseconds. ZT Scan 2.0 DIA methods were also tested for these loadings, tuned specifically for acquisitions at the indicated SPD method (see Table 1). Protein groups and precursors were compared to results previously generated on the ZenoTOF 7600+ system with commercially available Evosep 200 SPD, 300 SPD, and 500 SPD methods (3).
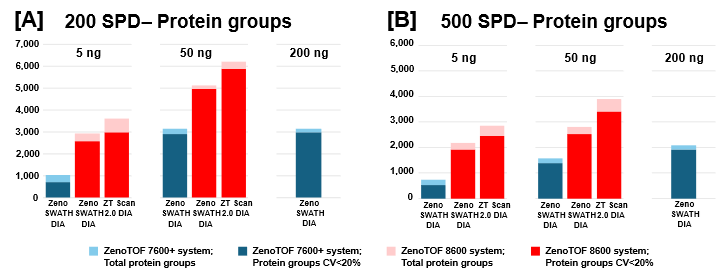
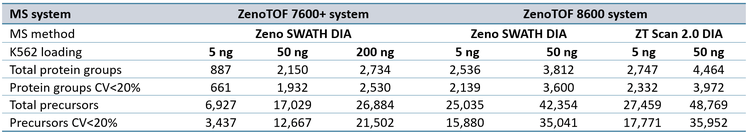
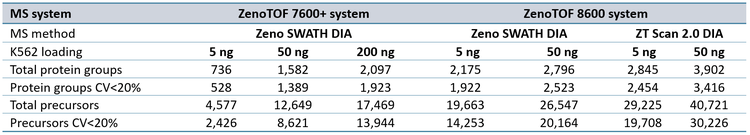
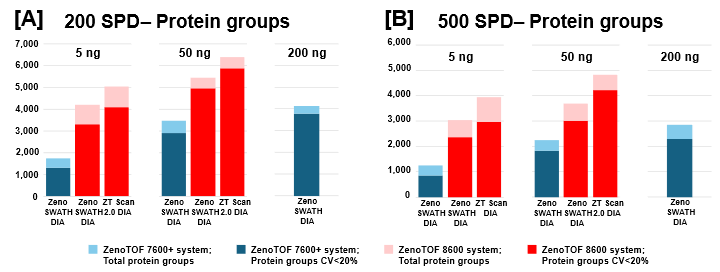
The protein group identifications from data processing with DIA-NN software are summarized in Figure 2. The graph clearly shows performance gains on the ZenoTOF 8600 system relative to the ZenoTOF 7600+ system, for both total protein groups and protein groups with CV <20% across replicates, at 5 ng and 50 ng loadings run with 200 or 500 SPD. The numbers of protein groups for all experiments are summarized in Supplemental Tables 1-3.
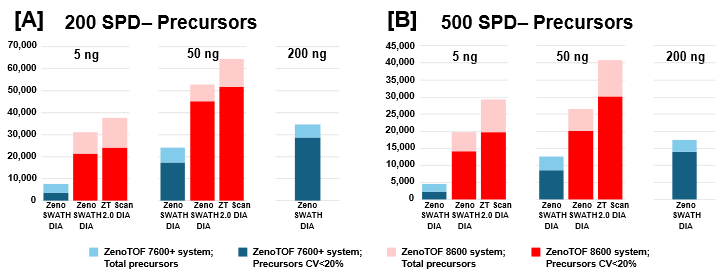
Similar improvements were observed for precursor identifications, summarized in Figure 3, whereby significant gains were achieved for both total precursors and those with CV <20% (see also Supplemental Tables 1-3).
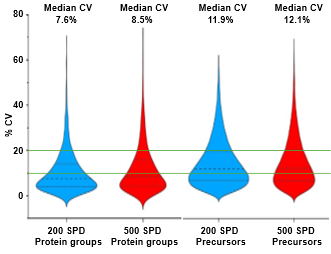
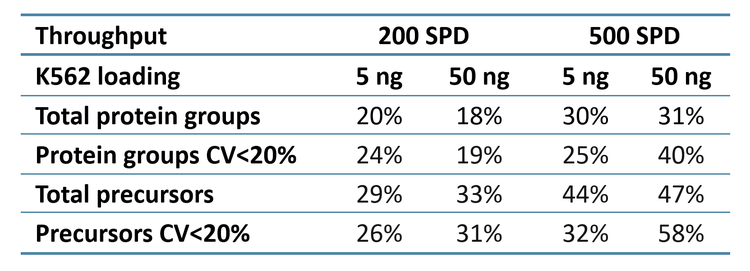
ZT Scan 2.0 DIA further improved total and quantitative identifications for both sample loadings and at both throughputs (Table 2). The enhancements were most pronounced at 500 SPD, where protein group gains were as much as 40%, and precursor gains as much as 58%. This trend of ZT Scan 2.0 DIA having increased benefits for combinations of higher loadings and throughputs correlates with previous observations on the ZenoTOF 7600+ system (3,4).The CV distributions for protein groups identified with ZT Scan 2.0 DIA on the ZenoTOF 8600 system are summarized in Figure 4.
Of note is the direct comparison between identifications at 200 ng loadings on the ZenoTOF 7600+ system to identifications at lower loadings on the ZenoTOF 8600 system. As shown in both Figures 2 and 3, along with Supplemental Tables 1-3, a higher number of identifications were observed on the ZenoTOF 8600 system at 5 ng loadings than with the ZenoTOF 7600+ system at 200 ng loadings, driven by the sensitivity gains of the ZenoTOF 8600 system.
Data files were also searched using PEAKS Studio software, resulting in fewer protein group identifications, but higher precursor identifications than with DIA-NN software (see Supplemental Figures 1 and 2, along with Supplemental Tables 4-6). Nevertheless, the same overall trends were observed as with the DIA-NN software processing results, namely (i) substantial improvements in total and quantitative protein groups and precursors using the ZenoTOF 8600 system relative to the ZenoTOF 7600+ system for a given loading and throughput, (ii) that ZT Scan 2.0 DIA improved the numbers of total and quantifiable protein groups and precursors over Zeno SWATH DIA on the ZenoTOF 8600 system, and (iii) that more protein groups and precursors were identified and quantified from 5 ng loads on the ZenoTOF 8600 system than from 200 ng loads on the ZenoTOF 7600+ system. The differences in the number of protein groups identified using DIA-NN software and PEAKS Studio software are presumably due to the differences in protein inference performed by the respective software algorithms. The higher number of precursors identified using PEAKS Studio software is likely due to the higher efficiency of identifications when doing a combination of spectral library and FASTA searches.
Sensitivity gains of the ZenoTOF 8600 system at the LC-MS peak and spectral level
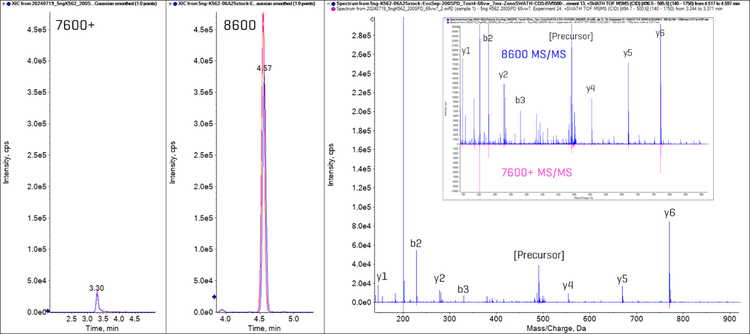
Figure 5 shows the sensitivity differences between the ZenoTOF 7600+ and 8600 systems for a representative peptide (DLTFYLMK, from protein cytoplasmic actin protein ACTB_HUMAN). The extracted ion chromatograms (XICs) for the y5 fragment ion from the 2+ charged precursor are shown from the Zeno SWATH DIA experiments for both the ZenoTOF 7600+ and 8600 systems. The XIC peak area is 10-fold higher on the ZenoTOF 8600 system. An overlaid comparison of the MS/MS spectra for this peptide also highlights the magnitude of the signal intensity differences across the fragment ion mass range between these systems.
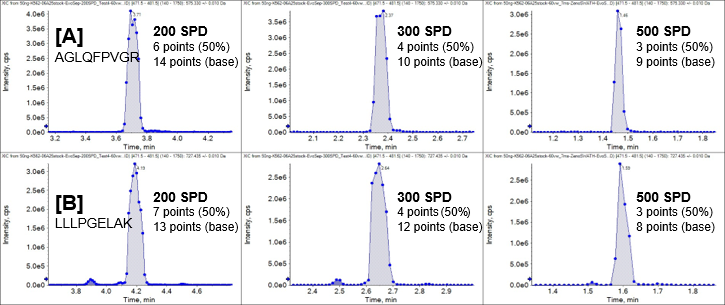
Quantitation is highly influenced by the quality of the LC-MS peak shape, which in turn is determined by the chromatographic separation. To examine this in greater depth, fragment ion XICs were examined for two peptides from the Zeno SWATH DIA experiments performed on the ZenoTOF 8600 system. Figure 6 shows XICs for peptide fragment ions H2A1C_HUMAN.AGLQFPVGR.2+.y5 and H2B1L_HUMAN.LLLPGELAK.2+.y7, acquired from 50 ng K562 digest, run at 200, 300, and 500 SPD. As expected, the number of data points at half-height decreases as the LC method gets faster due to the peak widths getting progressively narrower with sharper active gradients. However, even at 500 SPD throughput, at least 3 points across the chromatographic peaks at half-height are achieved. This demonstrates the high acquisition speeds of the DIA methods on the ZenoTOF 8600 system, enabling robust quantitation with high precision and accuracy.
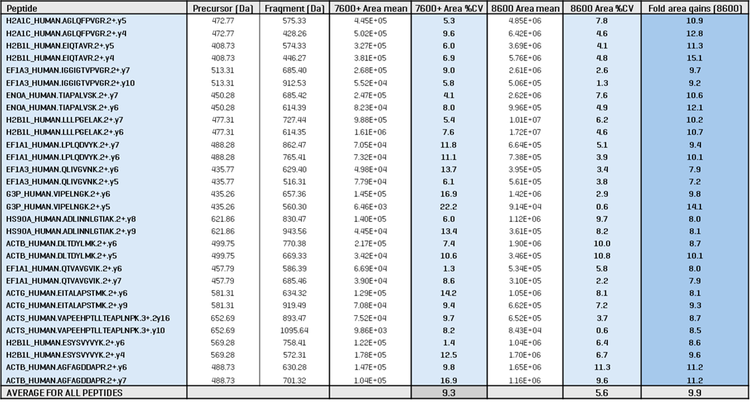
A panel of 15 peptides was selected for detailed quantitative analysis of the sensitivity gains with the ZenoTOF 8600 system (Table 3). For the 5 ng K562 loadings run at 200 SPD, two fragment ions were chosen for each peptide, and the XIC peak area differences were compared between the ZenoTOF 7600+ and 8600 systems. The reproducibility (i.e., % CV) between triplicate injections was also compared. The table shows that the average XIC peak area gain was 10-fold across this peptide panel and that the CVs between replicates improved from 9.3% on the ZenoTOF 7600+ system to 5.6% on the ZenoTOF 8600 system. This demonstrates the impact of higher sensitivity on the quantitative reproducibility of the results, which is an important consideration in terms of the overall data quality.
Conclusions
-
The ZenoTOF 8600 system identified >5x more precursors and >3x more protein groups, at sample throughputs of up to 500 SPD, relative to previous-generation QTOF systems.
-
The increased sensitivity of the ZenoTOF 8600 system resulted in more precursors and protein groups being identified in 5 ng sample loads than in 200 ng sample loads with the ZenoTOF 7600+ system.
-
Quantitative protein group and precursor identifications increased by as much as 58% using ZT Scan 2.0 DIA on the ZenoTOF 8600 system.
Acknowledgments
The authors would like to acknowledge Evosep (Denmark) for their contributions to this work.
References
-
Demichev et al., Nature Methods, 2020, https://www.nature.com/articles/s41592-019-0638-x
-
Large-scale protein identification using microflow chromatography on the ZenoTOF 7600 system. SCIEX technical note, RUO-MKT-02-14415-A.
-
Selecting optimal ZT Scan DIA acquisition methods on the ZenoTOF 7600+ system for quantitative proteomics. SCIEX technical note, MKT-34022-A.
-
Improved proteomics performance at high throughput using ZT Scan DIA on the ZenoTOF 7600+ system, SCIEX technical note, MKT-32367-A.