Abstract
This technical note summarizes a comparison of the 3 available ZT Scan data-independent acquisition (DIA) methods on the ZenoTOF 7600+ system for quantitative proteomics. ZT Scan DIA improves the identification and quantitation of peptides and proteins compared to discrete-window DIA methods by combining enhanced selectivity of the scanning quadrupole dimension with high sensitivity MS/MS using Zeno trap pulsing. ZT Scan DIA was tested across a range of sample throughputs and loadings using the Evosep One system and a complex lysate digest. These results provide guidance for users when choosing an appropriate ZT Scan DIA method for their proteomics experiments.
Key features of high throughput quantitative proteomics using ZT Scan DIA on the ZenoTOF 7600+ system
- ZT Scan DIA methods are pre-built and easy to set up, with minimal optimization required
- With high throughput microflow separations using the Evosep One system, ZT Scan DIA improves the number of identified protein groups and precursors by as much as 30% and the number of quantified protein groups and precursors by >50%
- Using these results as a guide, an appropriate ZT Scan DIA method can be selected for a specific gradient and on-column sample loading amount to maximize quantitative proteomics results
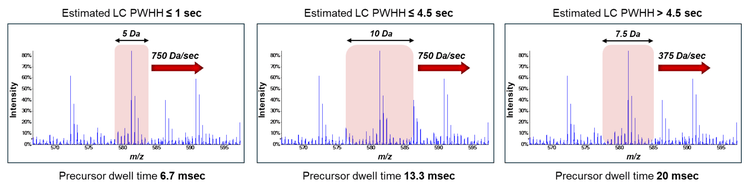
Introduction
DIA has become the acquisition method of choice for MS-based proteomics analysis, enabling the identification and quantitation of large numbers of proteins and peptides to enhance understanding of the complex biological mechanisms of various diseases. A critical aspect of disease research is analyzing large sample cohorts to increase the statistical significance of observed trends and better understand the disease mechanisms involved. As such, the ability to analyze large numbers of samples faster, with easy-to-implement methods and without compromising data quality, is of great value to researchers. The power of Zeno SWATH DIA for fast, high-quality quantitative proteomics has been previously demonstrated [1]. ZT Scan DIA has recently been presented as the next step in the evolution of DIA [2]. Using a scanning quadrupole coupled to fast, sensitive, time-of-flight (TOF) analysis improves specificity over traditional discrete-window DIA methods for accurately and precisely identifying and quantifying analytes across a given mass range. Performance improvements over Zeno SWATH DIA methods have been shown using pre-defined methods requiring minimal optimization and parameter setup [3,4]. This technical note details the selection of the ideal ZT Scan DIA method for identifying and quantifying the most protein groups and precursors in a complex lysate digest at various sample loads and throughputs
Methods
Sample preparation: K562 human cell lysate was acquired from Promega and reconstituted to the indicated concentrations with 0.1% formic acid in water.
Chromatography: Chromatographic separation was carried out using the Evosep One system (Evosep, Denmark) using preset methods with the flow rate, gradient length, overall throughput conditions, and Evosep columns specified in Table 1. Mobile phase A was water with 0.1% formic acid, and mobile phase B was acetonitrile with 0.1% formic acid. All data was acquired in triplicate.
Mass spectrometry: ZT Scan DIA analysis was performed on the ZenoTOF 7600+ system using the OptiFlow Turbo V ion source with the 1-10 µL/min microflow electrode. Evosep columns were connected to the microflow probe using a PEEK 1/16” high-pressure union (IDEX P/N P-779). The source parameters are listed in Table 2. The three available ZT Scan DIA methods, as described in Figure 1, were tested. For comparison, data was also acquired using Zeno SWATH DIA, using a method with 65 variable-width SWATH windows and an MS/MS accumulation time of 13 msec per SWATH window.
Data processing: All DIA data were processed using DIA-NN software version 1.9 [5]. A K562/HeLa spectral library previously described was used [6]. Default DIA-NN software search settings were used with the following changes: precursor m/z range was adjusted to 400-900, fragment m/z range was adjusted to 140-1750, MS1 accuracy was set to 0 ppm, MS2 accuracy was set to 20 ppm, scan window was set to 6, and Match Between Runs (MBR) was used. Quantification strategy was set to “Legacy” mode. The “--scanning-swath” command option was used when processing ZT Scan DIA data. Triplicate data files for a given load/gradient were searched together. The numbers of protein groups and precursors were determined from the output “pg_matrix.tsv” and “pr_matrix.tsv” files.

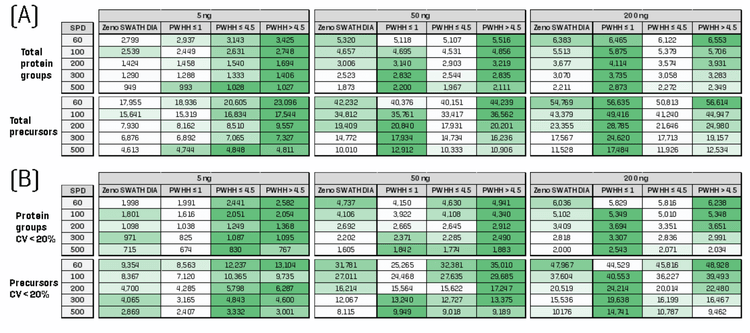

Comparison between ZT Scan DIA methods for quantitative proteomics
ZT Scan DIA methods are designed based on the estimated average LC peak width at half height (PWHH) (see Figure 1), which depends on LC column performance, flow rate, gradient length and gradient profile. Three on-column loadings of commercial K562 tryptic digest (5 ng, 50 ng, 200 ng) were run using 5 different samples-per-day (SPD) throughput methods with the Evosep One system (60, 100, 200, 300 and 500 SPD). The available ZT Scan DIA methods (PWHH ≤ 1 sec; PWHH ≤ 4.5 sec; PWHH > 4.5 sec) were tested for all sample loadings and throughputs, and the number of identified and quantified (i.e., CV < 20% across replicates) protein groups and precursors were compared. A standard Zeno SWATH DIA method was also run for comparison against each ZT Scan DIA method.
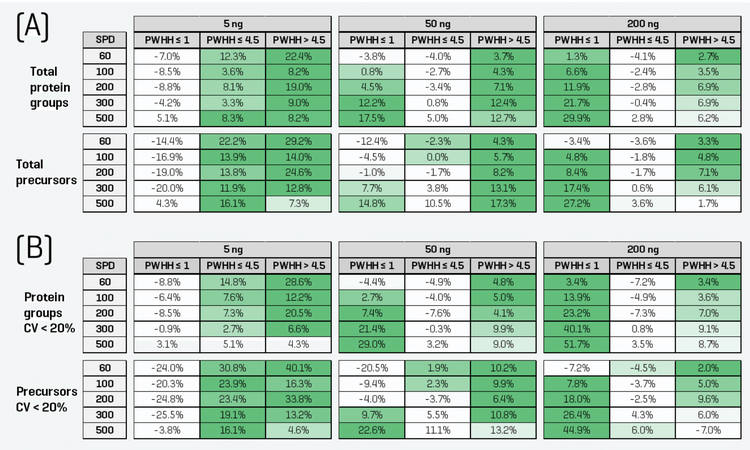
Figure 2 shows the total protein groups and precursors identified and quantified from each experiment. The color scale in the figure correlates to the performance magnitude for a given sample loading and throughput. The percentage gains for each ZT Scan DIA method relative to the reference Zeno SWATH DIA method are summarized in Figure 3, and the color scale again correlates to the magnitude of the gain for a given sample loading and throughput method. The results indicate that with the appropriate ZT Scan DIA method, improvements were observed relative to Zeno SWATH DIA at all loadings and throughputs. Interestingly, each ZT Scan DIA method displayed unique trends and characteristics regarding where it was most effective. The PWHH ≤ 1 sec method was most effective with faster gradients, particularly for higher on-column sample loadings, with gains in total identifications of as much as 30% and quantitative identifications of 52% (Figure 3). This is consistent with previous observations with high-throughput proteomics using ZT Scan DIA [3]. Therefore, this is the method of choice for high on-column loadings and high-throughput separations. The PWHH ≤ 4.5 sec method was most effective at low sample loadings, specifically with the number of quantitative identifications with CV < 20% across replicates. At higher sample loadings and throughputs, the effectiveness of this method diminishes. Finally, the PWHH > 4.5 sec generally showed higher effectiveness at low throughputs. This is not surprising, as it was designed to have the longest precursor dwell time and, therefore, should be most effective with broader LC peaks resulting from longer separation gradients. This method showed the best results when used at mid- to low sample loadings, and in many cases, its effectiveness surpassed that of the PWHH ≤ 4.5 sec method.
These findings highlight the gains that ZT Scan DIA provides for quantitative identifications over Zeno SWATH DIA. For users seeking to maximize proteomics performance when using high-throughput separation regimes on complex samples, these results guide method selection to help further streamline method development.
Conclusions
- For higher sample loadings and fast sample throughputs, the PWHH ≤ 1 sec ZT Scan DIA method works best, providing gains in total identifications of as much as 30% and gains in quantitative identifications of as much as 52%
- For mid- to low sample loadings and mid- to low throughputs, the PWHH > 4.5 sec ZT Scan DIA method was the ideal choice, showing gains in total identifications of as much as 29% and gains in quantifiable identifications of as much as 40%
- These results provide guidance for users when choosing an appropriate ZT Scan DIA method for a given proteomics experiment
References
- Flexibility, speed, and throughput for high proteome coverage using Zeno SWATH data-independent acquisition (DIA) coupled with the Evosep One system. SCIEX technical note, RUO-MKT-02-15461.
- Continuing the data-independent acquisition (r)evolution: Introducing ZT Scan DIA for quantitative proteomics. SCIEX technical note, MKT-31731-A.
- Improved proteomics performance at high throughput using ZT Scan DIA on the ZenoTOF 7600+ system, SCIEX technical note, MKT-32367-A.
- Improved proteomics performance or low sample loadings using ZT Scan DIA on the ZenoTOF 7600+ system, SCIEX technical note, MKT-32501-A
- Demichev et al., Nature Methods, 2020, https://www.nature.com/articles/s41592-019-0638-x
- Large-scale protein identification using microflow chromatography on the ZenoTOF 7600 system. SCIEX technical note, RUO-MKT-02-14415-A.