Comprehensive characterization of O-linked glycosylation in etanercept by electron activated dissociation (EAD)
Featuring the ZenoTOF 7600 system and Biologics Explorer software from SCIEX
Haichuan Liu1, Xuezhi Bi2 and Zoe Zhang1
1SCIEX, USA
2Bioprocessing Technology Institute (BTI), Singapore
Introduction
This technical note highlights the power of electron activated dissociation (EAD) for confident identification and unambiguous localization of O-linked glycosylation in etanercept. The unique ability of EAD to pinpoint the positions of glycosylation further enabled differentiation of positional isomers of O-glycopeptides.
Glycosylation is a common post-translational modification (PTM) that plays a critical role in antibody effector functions.1 Comprehensive characterization of N- and O-linked glycosylation in protein therapeutics is essential for ensuring drug safety and efficacy.1 When applied to glycopeptides, traditional collision-based MS/MS approaches, such as collision induced dissociation (CID), result in the loss of labile glycan moieties. Hence, accurate determination of glycosylation sites using CID is extremely challenging, particularly for O-glycosylation without a consensus sequence. Compared to CID, EAD is superior in glycopeptide analysis given its ability to preserve the glycan structures in the fragments.2–4
Etanercept is a dimeric fusion protein consisting of two tumor necrosis factor receptor (TNFR)-Fc chains with 3 N-glycosylation and 13 O-glycosylation sites on each chain. Glycosylation of etanercept was characterized on the subunit levels in previous technical notes.5 In this work, EAD was utilized to elucidate the complex O-glycosylation profile on the peptide level.
Key Features
Key features of the SCIEX solution for in-depth characterization of glycosylation
- Accurate localization of glycosylation using EAD: Labile glycans can be retained in the EAD fragments, allowing unambiguous determination of glycosylation sites and confident differentiation of positional isomers of O-glycopeptides (Figure 1).
- Excellent sensitivity and high quality of MS/MS: The Zeno trap of the ZenoTOF 7600 system provides a 5–10-fold increase in detection of MS/MS fragments, leading to excellent MS/MS sensitivity and spectral quality.
- Fast and flexible: EAD can be operated in data-dependent acquisition (DDA) or MRMHR mode with fast scanning rate (~20 Hz in DDA mode) and with the ability to tune the electron kinetic energy (KE).
- Powerful data analysis platform: Biologics Explorer software offers powerful, intuitive tools for peptide mapping and PTM analysis.
Figure 1. Elucidation of O-linked glycosylation in etanercept using the ZenoTOF 7600 system and Biologics Explorer software from SCIEX. EAD is powerful for localization of labile glycosylation. The sites of glycosylation in O-linked glycopeptides of etanercept containing up to 7 O-linked glycan structures were confidently determined based on interpretation and annotation of EAD MS/MS spectra using Biologics Explorer software.
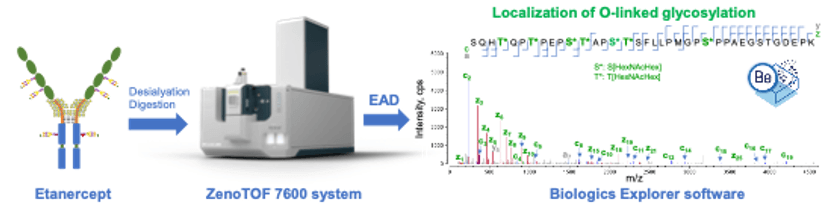
Methods
Sample preparation: Etanercept (25 μg/μL) was denatured by guanidine hydrocholoride (7 M), reduced with dithiothreitol (10 mM) and alkylated using iodoacetamide (25 mM). The sample solution was buffer exchanged into 50 mM Tris-HCl (pH = 7.4) using Bio-Spin columns (Bio-Rad), followed by enzymatic digestion at 37ºC for 2 h using trypsin (Promega). The resulting solution was incubated with SialEXO from Genovis at 37ºC for 4 h to remove sialic acids. A total of 10–20 μL of the final solution (~5–10 μg) was injected for LC-MS analysis.
Chromatography: The peptides were separated with an LC gradient, displayed in Table 1, using a Waters ACQUITY CSH C18 column (2.1 × 150 mm, 1.7 μm, 130 Å). A flow rate of 0.25 mL/min was used for the separation. The column was kept at 60ºC in the column oven of an ExionLC system from SCIEX. The mobile phases A and B consisted of 0.1% formic acid (FA) in water and 0.1% FA in acetonitrile, respectively.
Table 1. LC gradient.
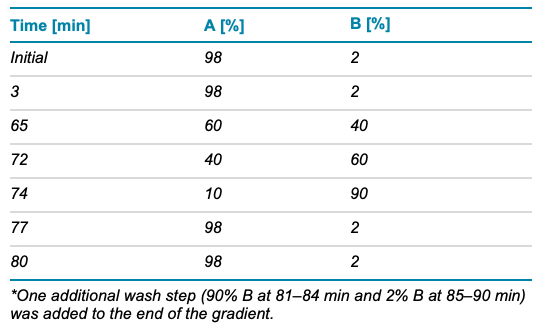
Table 2. TOF MS parameters.
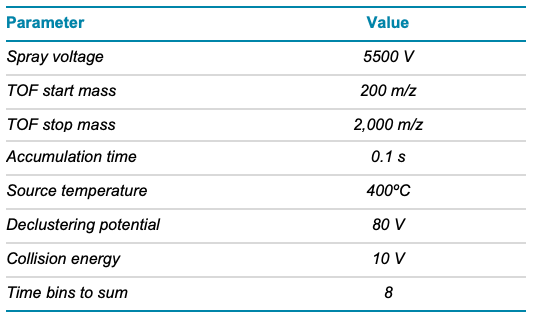
Mass spectrometry: EAD DDA and MRMHR data were acquired in SCIEX OS software using the ZenoTOF 7600 system. EAD MRMHR was specifically applied to O-linked glycopeptides containing 6 or 7 core 1 O-glycans. The key TOF MS and EAD (DDA or MRMHR) settings are listed in Tables 2–4.
Table 3. EAD DDA parameters.
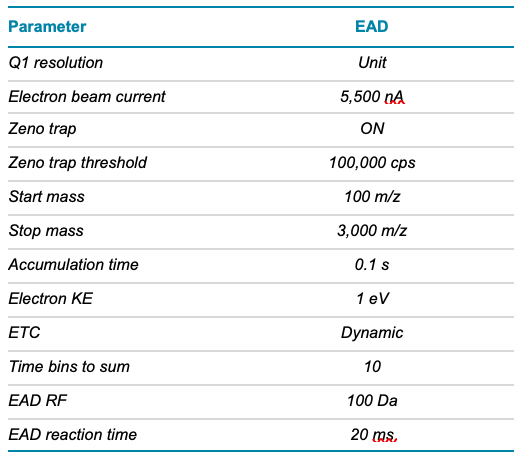
Table 4. EAD MRMHR parameters.

Data processing: The EAD DDA and MRMHR data were analyzed using two peptide mapping templates within the Biologics Explorer software.
Glycosylation in etanercept
Etanercept consists of 3 N-glycosylation and 13 O-glycosylation sites on each TNFR-Fc chain. Most of the O-glycosylation sites are located in the hinge region. Comprehensive characterization of glycosylation using collision-based MS/MS approaches, such as CID, is challenging because these methods cannot provide site-specific information about glycosylation due to the loss of labile glycans. By contrast, EAD has the unique ability to preserve the glycans in the fragments,2–4 allowing unambiguous localization of this labile PTM. In this technical note, the EAD DDA or MRMHR method was employed to characterize the complex O-glycosylation profile of desialylated etanercept.
Glycopeptides containing 1 O-glycan
Figure 2 displays EAD spectra of two tryptic O-glycopeptides (L1-R19 and T243-K268) containing a core 1 glycan structure (HexNAcHex). Both peptides contain 3 potential O-glycosylate sites (2 Thr and 1 Ser), but only one of the sites was modified with HexNAcHex. As shown in Figure 2, EAD generated excellent quality MS/MS spectra with a nearly complete series of c/y/z fragments, leading to confident identification of these two O-glycopeptides and unambiguous localization of the glycan. Specifically, the O-glycosylation site in peptide L1-R19 (Figure 2A) was determined to be T8 based on the m/z of c7/c9 and y11/z12 fragments, while T245 in peptide T243-268 was modified with HexNAcHex based on the detection of the non-glycosylated c2/y23 and glycan-containing c3/y24/z24.
Figure 2. EAD spectrum (1 eV) of singly O-glycosylated peptides L1-R19 (A) and T243-K268 (B). Two tryptic peptides contain multiple O-glycosylation sites (Ser and Thr), one of which was modified with the core 1 glycan structure (HexNAcHex). The excellent EAD MS/MS spectra allowed for confident localization of the O-glycan at T8 in peptide L1-R19 (A) and at T245 in peptide T243-K268 (B). Note that all the EAD spectra displayed in this technical note are deisotoped spectra taken from Biologics Explorer software. Not all fragments were labeled for spectral clarity.
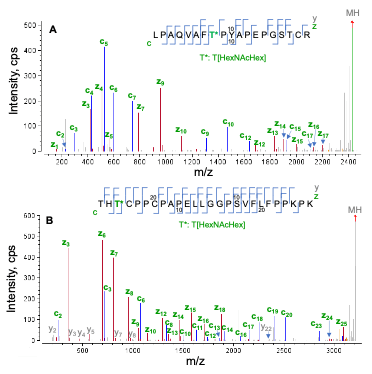
Figure 3. Characterization of 3 isomers of the singly O-glycosylated peptide SMAPGAVHLPQPVSTR (S186-R201) using EAD (1 eV). Three chromatographic peaks were detected in the extracted ion chromatogram (XIC) of S186-R201 modified with 1 HexNAcHex (A). The high-quality EAD data (B–D) revealed that two (S199 and T200) out of three potential O-glycosylation sites in this peptide were occupied. Based on the m/z of z2 and z3, the peak at RT = 22.6 min (A) was associated with O-glycosylation at S199 (D) while both peaks at RT = 20.8 min and 21.6 min can be assigned as the species carrying 1 HexNAcHex at T200 (B and C).
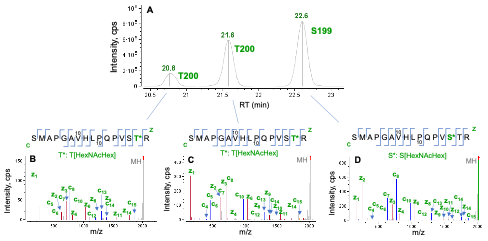
Figure 3 shows an example of EAD for differentiation of positional isomers of a singly O-glycosylated peptide (S186-R201). Three isomeric species were observed in the XIC of O-glycopeptide S186-R201 (Figure 3A). The high-quality EAD data revealed that the most abundant isomer detected at RT = 22.6 min corresponded to O-glycosylation at S199 (Figure 3D) while the other two species (RT = 20.8 min and 21.6 min) were modified with 1 HexNAcHex at T200 (Figure 3B and 3C). The presence of two T200 isomers might be attributed to different linkages of the glycan. This result highlights the unique capabilities of EAD for confident identification of O-glycopeptides, accurate localization of glycosylation and isomer differentiation in one experiment.
Glycopeptides containing multiple O-glycans
The EAD MS/MS spectra of O-glycopeptide S186-R201 modified with 2 or 3 HexNAcHex are displayed in Figure 4. For the doubly O-glycosylated species (Figure 4A), the detection of a nearly complete series of c/z fragments with or without the glycan enabled accurate localization of 2 HexNAcHex at S199 and T200. In the case of triply glycosylated species, the m/z of c series ions confirmed that the Ser residue at N-terminus was also occupied by HexHAcHex (Figure 4B), as expected.
Figure 4. EAD MS/MS spectra (1 eV) of the O-glycopeptide S186-R201 modified with 2 (A) or 3 (B) HexNAcHex. EAD produced a nearly complete series of c/z fragments for unambiguous localization of multiple HexNAcHex in this O-glycopeptide.
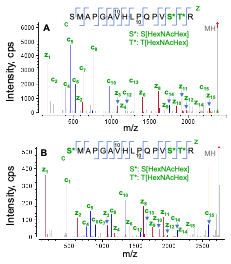
Figure 5. EAD MS/MS spectra (7 eV) of the O-glycopeptide S202-K238 modified with 6 (A) or 7 (B) HexNAcHex. The O-glycopeptide S202-K308 contains up to 7 O-glycans. Accurate localization of the O-glycans was achieved for the species containing 6 (A) or 7 (B) HexNAcHex using EAD. Note that the EAD MS/MS spectrum of one of two positional isomers for the species carrying 6 HexHAcHex is displayed in Figure 5A. See Figure 6 for differentiation of two isomers.
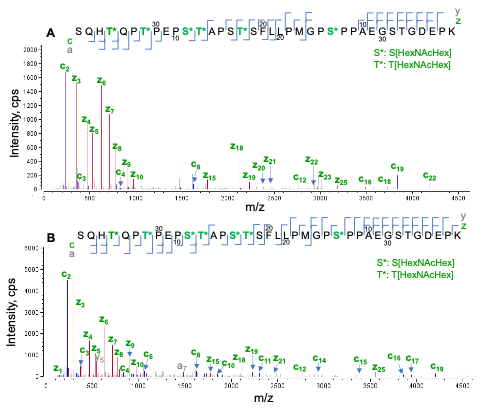
Trypsin digestion of etanercept generated a long tryptic peptide (S202-K238) that contains 11 potential O-glycosylation sites (6 Ser and 5 Thr). EAD data confirmed that 7 out of 11 of these sites were occupied with the O-glycans. Figure 5 displays the EAD MS/MS spectra of the species containing 6 or 7 HexNAcHex. The presence of multiple proline residues (9 in total) prevented the generation of c/z fragments that correspond to the cleavages at the N-termini of the proline. This limitation was partially overcome by the detection of additional a/y fragments at KE = 7 eV. By taking all the sequence ions (c/z/a/y) into consideration, the locations of 6 or 7 HexNAcHex can be confidently pinpointed in the sequence (Figure 5).
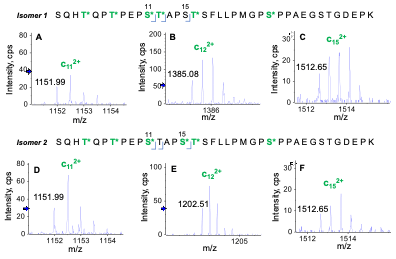
Two positional isomers were observed for glycopeptide S202-K238 containing 6 HexNAcHex moieties. Figure 6 shows the signature EAD fragments—i.e., doubly charged c11, c12 and c15 for differentiation of these two isomers. The m/z difference (365 Da) seen for c12 produced from EAD of two isomers indicated that T213 (T12 in the sequence) was O-glycosylated in Isomer 1 (Figure 6B) but not in Isomer 2 (Figure 6E). The fact that the m/z of c15 was the same for two isomers showed that S216 (S16 in the sequence) was modified with a HexNAcHex in Isomer 2 (Figure 6F) but non-glycosylated in Isomer 1 (Figure 6C). These results highlight the power of EAD for unambiguous differentiation of positional isomers, which is challenging for traditional collision-based MS/MS approaches, such as CID.
Figure 6. Signature c ions for differentiation of two positional isomers of the O-glycopeptide S202-K238 containing 6 HexNAcHex. Two isomers (Isomer 1 and Isomer 2) were differentiated based on the detection of c11 2+ and c15 2+ ions at the same m/z (A and D) and a c12 2+ fragment containing 4 and 3 HexNAcHex for Isomer 1 (B) and Isomer 2 (E), respectively. The m/z of c12 2+ indicated that T12 in the sequence was modified with a HexNAcHex in Isomer 1 (B) but not in Isomer 2 (E), where S15 was glycosylated based on the m/z of c15 2+ (F).
Conclusion
- Confident identification and accurate localization of Oglycosylation in etanercept were achieved.
- High-quality EAD data enabled localization of O-glycopeptides containing up to 7 O-glycans.
- Positional isomers of O-glycopeptides were differentiated unambiguously based on a complete or nearly complete series of sequence ions produced in EAD.
- The EAD approach described in this technical note can be leveraged to elucidate complex glycosylation profiles in protein therapeutics.
References
- Zhang P et al. (2016) Challenges of glycosylation analysis and control: an integrated approach to producing optimal and consistent therapeutic drugs. Drug Discovery Today 21(5):740-765.
- A new electron activated dissociation (EAD) approach for comprehensive glycopeptide analysis of therapeutic proteins. SCIEX technical note, RUO-MKT-02-12980-A.
- Comprehensive glycopeptide analysis of a protein-based vaccine. SCIEX technical note, RUO-MKT-02-13816-A.
- Site-specific N-linked glycan profiling on the fusion protein aflibercept using a novel fragmentation technique. SCIEX technical note, RUO-MKT-02-14145-A.
- Comprehensive characterization of etanercept glycosylation by subunit LC-MS analysis. SCIEX technical note, RUOMKT-02-13712-A.