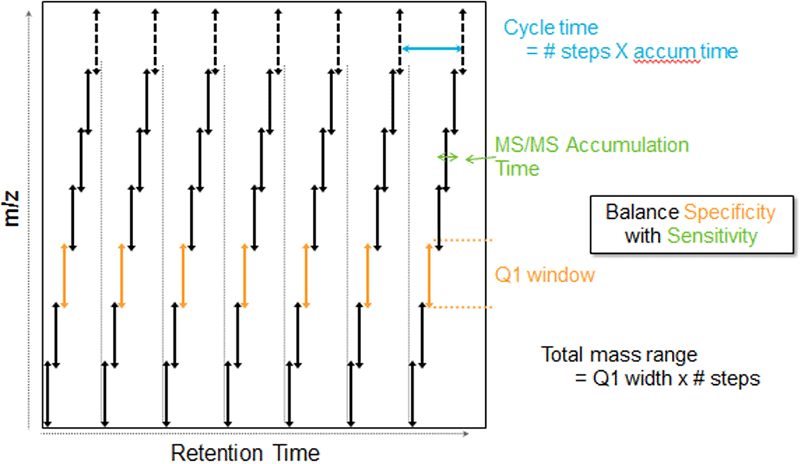
Since its release in 2012, SWATH DIA (data independent acquisition) has been a valuable tool for large-scale proteomic studies. SWATH DIA helps generate a permanent digital proteome map with highly reproducible retrospective analysis of cellular and tissue specimens. It combines quantitative reproducibility with the speed and sensitivity gains of modern mass spectrometers.
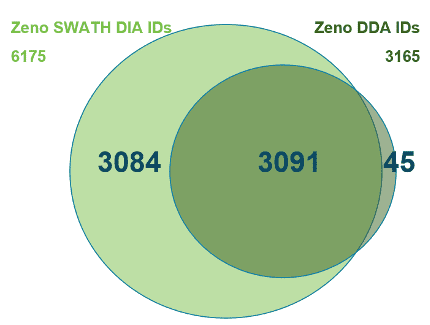
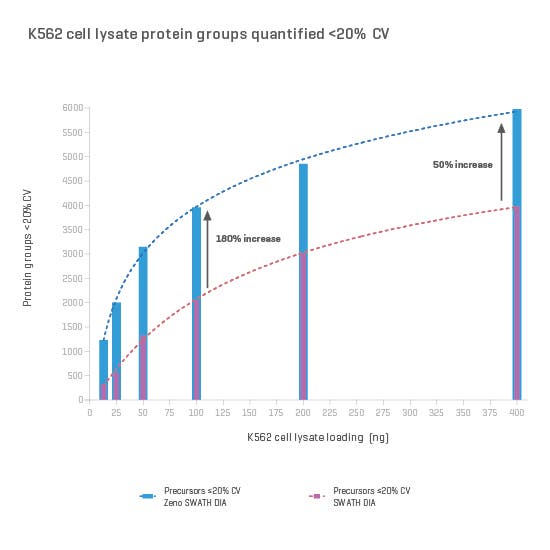
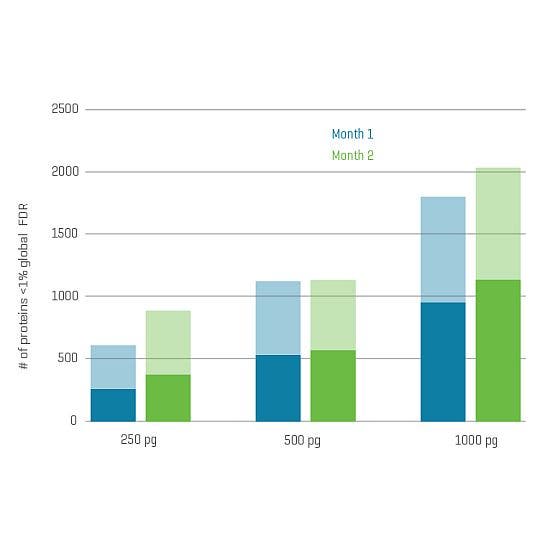
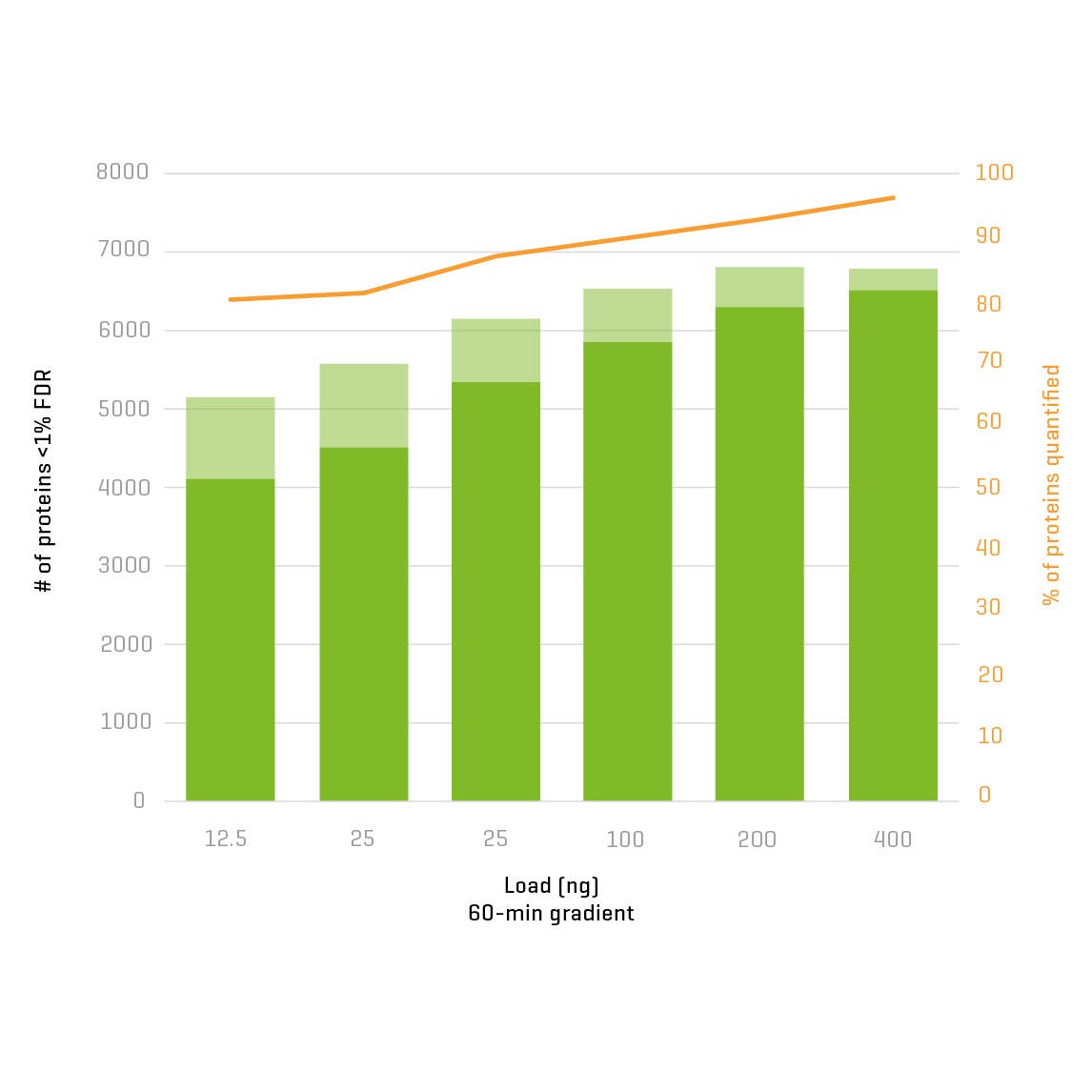
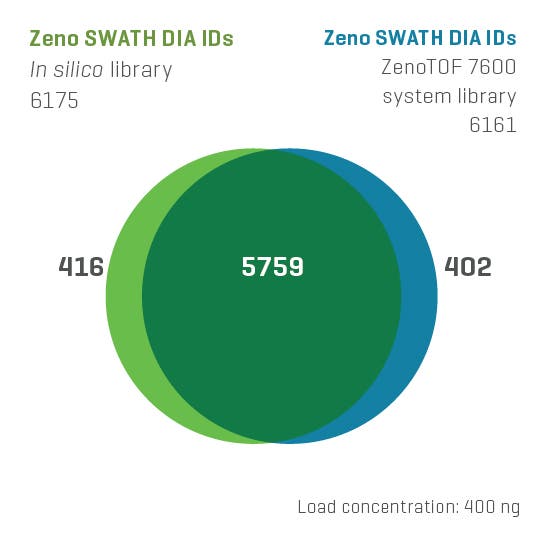
And now, with the support of powerful new hardware advancements, Zeno SWATH DIA reveals tens, hundreds and even thousands more identified and quantified analytes in less time and with higher precision than ever before. Reaching new depths of coverage, Zeno SWATH DIA delivers maximal information in minimal time.