Streamlined LC/MS analysis on crude bioreactor antibody samples using inline tryptic digest
Ronan Gough1, Vicky Smith1 and Stephen Lock2
1CPI, UK; 2SCIEX, UK
Abstract
This technical note demonstrates a feasibility study of an inline tryptic digestion prior to LC-MS analysis to reduce the time from question to answer. This workflow enables the analysis of crude bioreactor samples with simple sample preparation within an hour of sampling and facilitates quick critical quality attribute (CQA) identification and monitoring.
Introduction
Monoclonal antibodies (mAbs) are one of the most successful biotherapeutics used for the treatment of autoimmune diseases and various cancers.1 Production of mAbs involves cell cultures in either batch or continuous manufacturing. Process analytical technology (PAT) methods2 are frequently used to monitor and assess protein products and processes, aiming for rapid analysis of critical process parameters that impact CQAs.2
Mass spectrometry (MS) has often been applied to samples that have undergone prior cleanup. Typically, crude bioreactor samples require an extensive and time-consuming cleanup step before MS analysis can be conducted.3
This step is especially important for applications such as peptide mapping. Intensive sample preparations involving protein denaturation, reduction and alkylation are performed before the digestion and analysis of the sample. Sample preparations such as this are labor-intensive and time-consuming.4
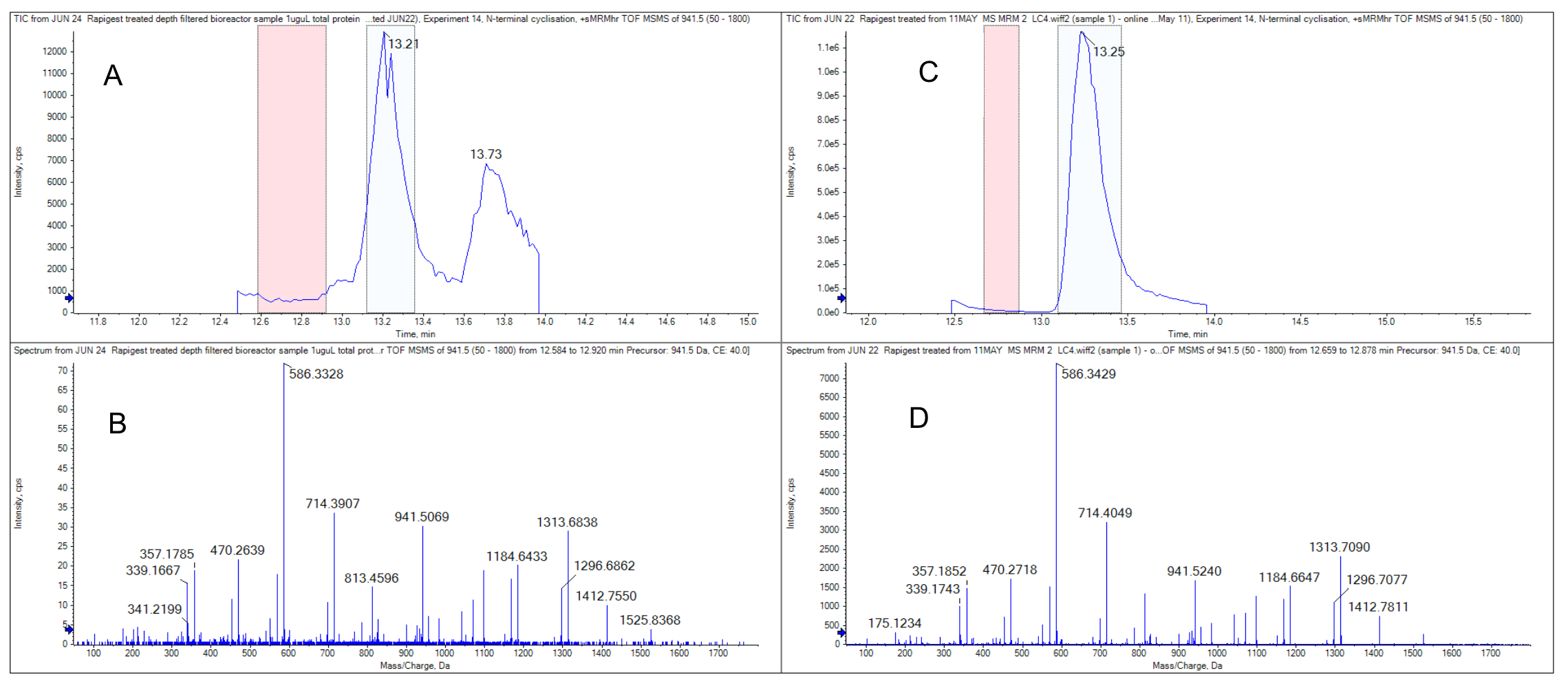
Figure 1. A comparison of a crude bioreactor sample and a pre-purified sample analyzed by inline, automated trypsin digestion. The results shown here highlight the detection of an N-terminal modification site within an hour. (A) An extracted ion chromatogram (XIC) for the N-terminal modified peptide from the bioreactor sample. (B) A product ion scan for the N-terminal peptide from the bioreactor sample. These data are from the peak at 13.21 minutes (blue region), which was background subtracted using the area around 12.5 minutes (red region). (C) An XIC for the N-terminal modified peptide from the pre-purified sample. (D) A product ion scan for the N-terminal peptide from the pre-purified sample. These data are from the peak at 13.21 minutes (blue region), which was background subtracted using the area around 12.5 minutes (red region).
Key features of inline tryptic digestion LC-MS
- Direct analysis of crude bioreactor samples: Minimal sample preparation is needed for the LC/MS analysis
- Inline enzymatic digestion: Automate trypsin digestion to reduce sample preparation time and streamline the workflow
- Fast turnaround time: Detection of target CQAs within 1 hour
Methods
Sample preparation: Bioreactor samples (950 µL from a batch process) were centrifuged (1000 g, 10 minutes) to remove cell debris. The sample was then mixed with 0.1% RapiGest (Waters) in 50mM Tris buffer (pH 8) and heated (15 minutes, 80°C). The sample was cooled to room temperature before injection.
Chromatography: Samples were separated using an ExionLC system that included a pair of ExionLC AD pumps (SCIEX, P/N: 5036653), an ExionLC AC pump (SCIEX, P/N: 5036649) and an ExionLC UV detector (SCIEX, P/N: 5036652).
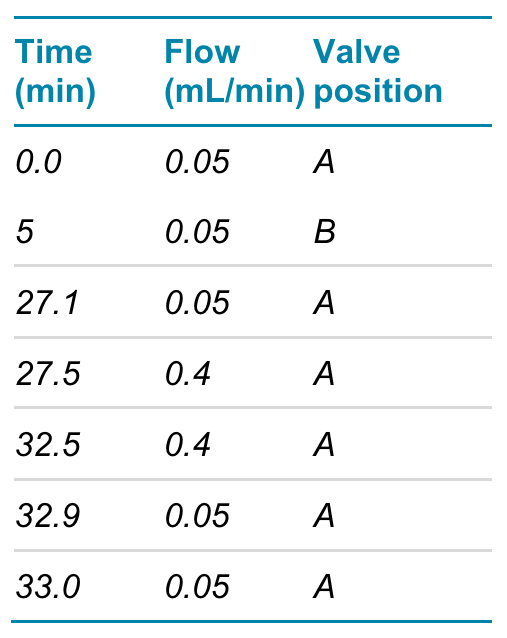
During sample injection and online tryptic digestion, the Perfinity trypsin column together with an ACQUITY UPLC BEH C18 VanGuard Pre-column, 130Å, 1.7 µm, 2.1 mm X 5 mm (used to capture the tryptic peptides) was plumbed into the inbuilt 6-port switching valve on the MS system. The LC flow exiting the trypsin column was directed to waste. After 5 minutes, the valve position was switched and the peptides were eluted and separated using a Synergi™ Fusion-RP C18 column (2.0 x 50 mm, 200 µL/min, Phenomenex). The elution gradient used 0.1% formic acid in water (mobile phase A) and 0.1% formic acid in acetonitrile (mobile phase B) at a column oven temperature of 37°C (see Tables 1 and 2 below).
Table 1. LC conditions used to separate peptides.
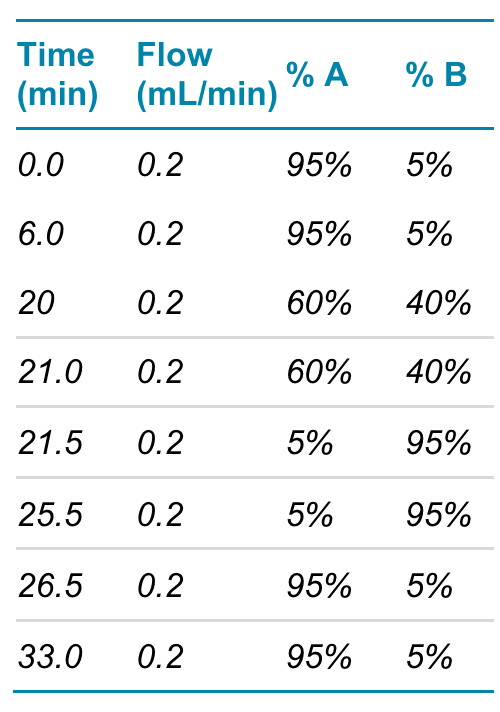
Table 2. Loading pump flow and switching valve position.
Mass Spectrometry: The mass spectrometer used for this proof-of-principle project was an X500B QTOF system (SCIEX). Twenty-five targeted product ion experiments were acquired using a scheduled method (MRMHR) to detect a subset of tryptic peptides. The TOF MS/MS spectra were acquired over a range of 450 to 2500 amu (accumulation time 0.25 s) using an ion spray voltage of 5500 V, source temperature of 350°C, declustering potential (DP) voltage of 80 V and collision energy (CE) of 40 V. The curtain gas and ion source gases 1 and 2 were set to 35 psi. The first peptide peak eluted at 6.75 minutes and the last peptide peak was detected at 16.45 minutes during the gradient ramp.
Data processing: Data collected using data-dependent acquisition (DDA) was analyzed using the Explorer module in SCIEX OS software.
Results and discussion
To prove the project concept, the LC methods were set up with a back pressure threshold under 2500 psi. This threshold was the pressure limit of the Perfinity trypsin column and was applied for the entire HPLC separation. Before a bioreactor sample test, purified trastuzumab (test sample) was digested using the classical in-solution digestion and tested. The digested trastuzumab was injected into the HPLC and it was confirmed that tryptic peptides were successfully trapped on the vanguard C18 trap column. After the elution, peptides were separated by column and detected by the X500B QTOF system. In this experiment, a platform peptide mapping method was used with a DDA experiment to confirm the identity of the target peptide (Figure 1).
Figure 2. The pre-digested sample was analyzed using a DDA method. A) The chromatogram of the triggered product ions. B) The XIC of the N-terminal HT 01 peptide4. C) The MS/MS spectra for the corresponding peptide, which is part of the variable domain of trastuzumab.
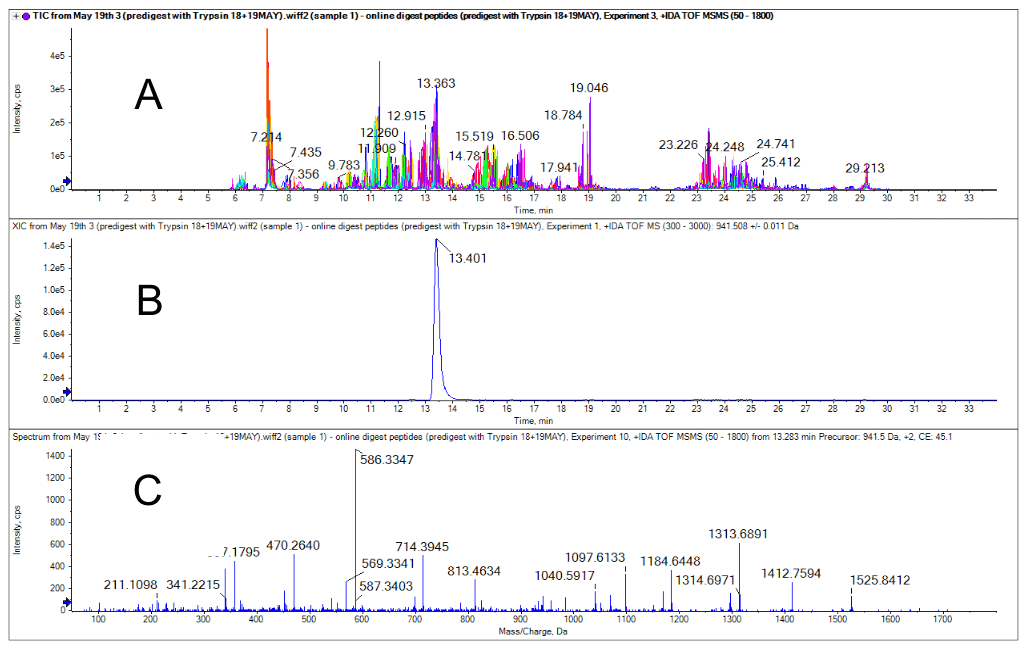
In Figure 2A, the dependent scans triggered from the tryptic peptides eluted between 6 and 20 minutes. Figures 2B and 2C show an example of the XIC for the precursor with m/z 941.508 and its corresponding MS/MS spectra. The peptides that eluted after 20 minutes come from a chemical background that elutes with the column wash step. Peptides of interest were identified from DDA experiments and the MRMHR experiment was designed to closely investigate a subset of 25 peptides from the 79 theoretical tryptic peptides from the trastuzumab digest. The MRMHR method was used to compare an undigested purified protein sample and a bioreactor sample. Figure 3 demonstrates an example comparison of the N terminal peptide from the 3 different analyses.
The results show that an identical product ion scan was obtained from the pre-digested standard, inline digested standard and the bioreactor sample. Similar retention times were observed in all 3 samples, however, the bioreactor sample had significantly higher background and its peak intensity was lower.
Figure 3. Comparison of the N-terminal peptide from the heavy chain (HC 01) region.4 A) the pre-digested sample, B) the inline digested pure standard and C) the bioreactor sample.
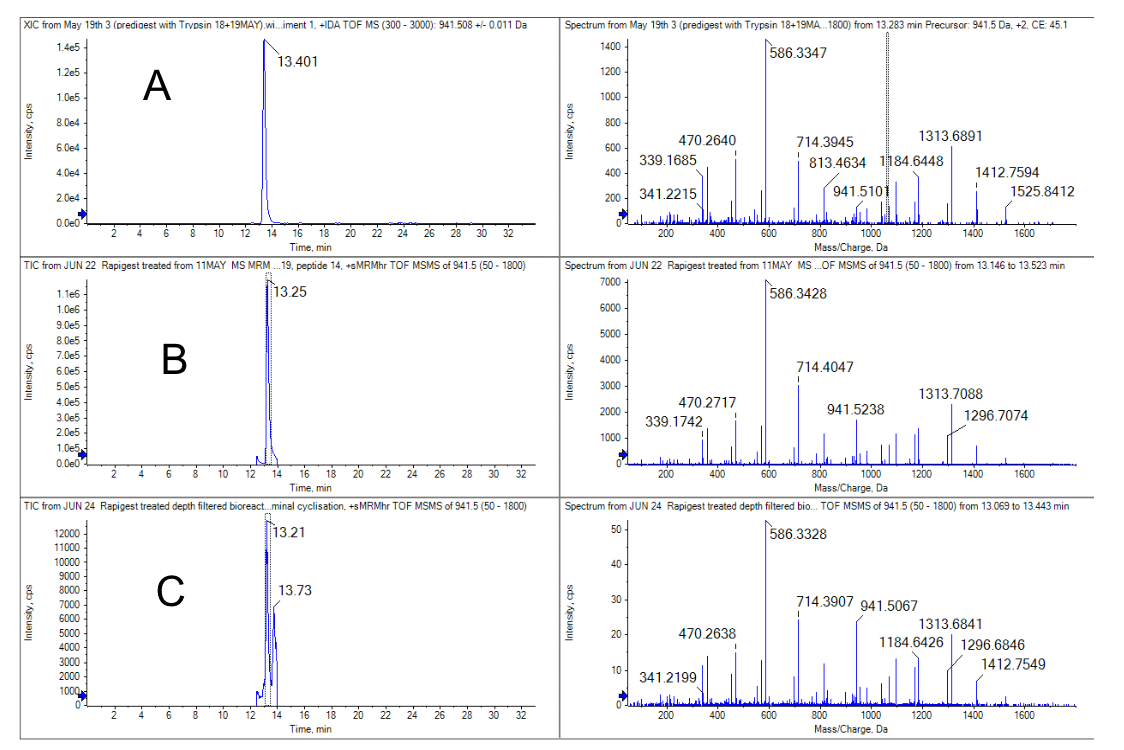
With similar injection volumes and protein loads, the response of the bioreactor sample was approximately 10-fold lower. Despite its attenuated response, the N-glycosylated peptides were still confidently detected and identified, as shown in Figure 4.
Figure 4. Comparison of the N-glycosylated peptide from the heavy chain (HC 29 – G0F) region.4 A) a pure standard and B) the bioreactor sample.
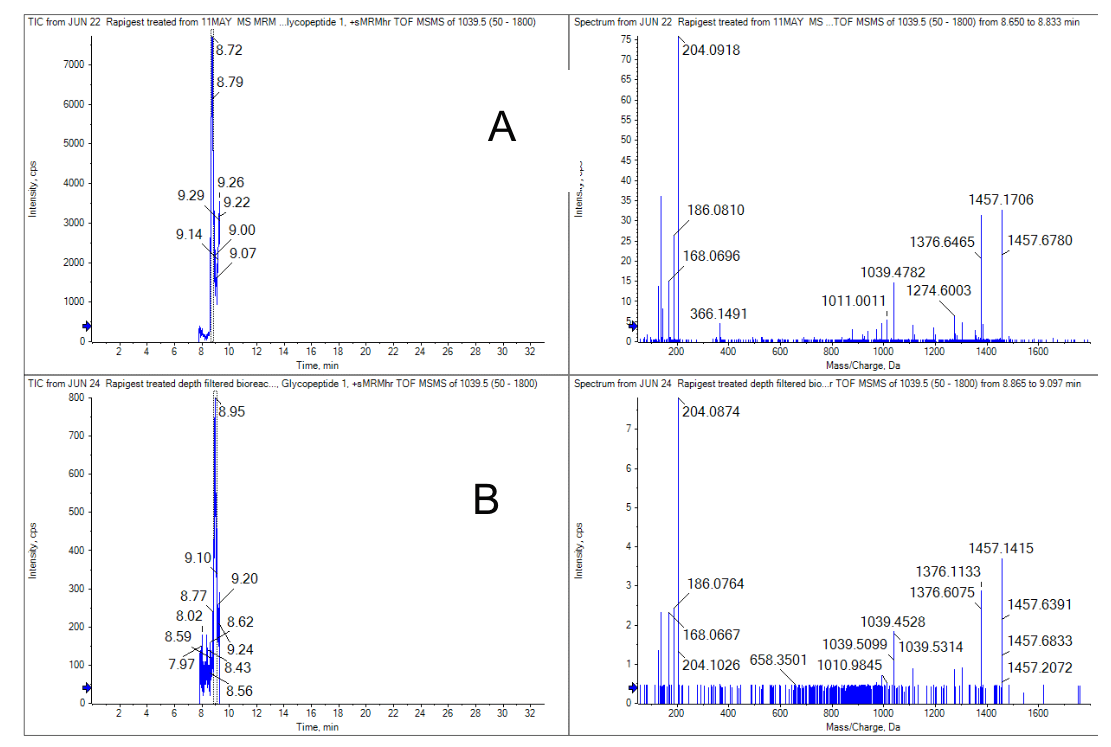
Small polar peptides were not selected for the MRMHR experiment, as they typically do not contain modification sites and would not be retained well on the C18 trap cartridge after digestion. The peptides with known modifications and sites of variability were targeted for this study.
Conclusion
- Inline tryptic digestion allows direct analysis of bioreactor samples to confidently identify peptides of interest and CQAs
- The method enables the identification of modifications, including N-glycan profiling, in less than an hour
- MS/MS methods are necessary to overcome the higher background of bioreactor matrix samples, as they provide higher specificity and sensitivity, leading to identifications with higher confidence
References
- Lu R. et al. (2020), Development of therapeutic antibodies for the treatment of diseases, Journal of Biomedical Science, Volume: 27, Article number: 1.
- Chopda V. et al. (2021), Recent advances in integrated process analytical techniques, modeling, and control strategies to enable continuous biomanufacturing of monoclonal antibodies. Journal of Chemical Technology & Biotechnology Volume 97, Issue 9, Pages 2317-2335.
- Rathore D. et al. (2018), The role of mass spectrometry in the characterization of biologic protein products, Expert Review of Proteomics, Volume: 15, Issue: 5, Pages: 431-449.
- Gahoual, R. et al. (2013), Rapid and multi-level characterization of trastuzumab using sheathless capillary electrophoresis-tandem mass spectrometry, mAbs, Volume 5, Issue 3, Pages 479-490.
- Andjelković, U. et al. (2017), Use of monolithic supports for high-throughput protein and peptide separation in proteomics, Electrophoresis, Volume 38, Issues 22-23, Pages 2851-2869.