Methods
Sample preparation: The mAb sample (adalimumab) was denaturated with 7.2M guanidine hydrochloride, 100mM Tris buffer pH 7.2, followed by reduction with 10mM DL-dithiothreitol and alkylation with 30mM iodoacetamide. Digestion was performed with trypsin/Lys-C enzyme at 37°C for 16 h.
Chromatography: 10 µL (4 µg) of the trypsin/Lys-C digest were separated with a CSH C18 column (2.1×100 mm, 1.7 μm, 130 Å, Waters) using an ExionLC system. The mobile phase A consisted of water with 0.1% formic acid, while the organic phase B was acetonitrile 0.1% formic acid. A gradient profile was used at a flow rate of 300 μL/min (Table 1). The column temperature was maintained at 50°C.
Mass spectrometry: Data were acquired in positive ionization mode with an information dependent acquisition (IDA) method using the SCIEX ZenoTOF 7600 system. The electron energy for the alternative fragmentation in the EAD cell was set to a value of 7 eV. Detailed method parameters are summarized in Table 2.
Data processing: Data were processed using Byos software (Protein Metrics Inc.).
The what, why and how
The formation of isoAsp has two routes: one is the deamidation of asparagine (Asn) and the other is the isomerization of Asp (Figure 2).4 The formation of isoAsp is a non-enzymatic process through a succinimide intermediate, usually increasing with prolonged storage time. Since the isomerization of Asp introduces an additional methylene group in the protein backbone, it can lead to a dramatic impact on the protein folding5 , which can affect the drug’s efficacy and safety. It was reported that isomerization of Asp in the complementary determining regions (CDRs) of antibodies decreased binding efficiency.6 PTMs with such an effect on the product are classified as critical quality attributes (CQA) and require comprehensive characterization and monitoring. Bottom-up approaches are the method of choice for characterization of product quality attributes, enabling the simultaneous identification and localization of modifications. Peptide mapping analyses with collision-induced dissociation (CID) are commonly utilized to investigate deamidation sites, as each deamidation will introduce a 0.98 Da mass shift. However, it is a challenge to distinguish isoAsp and Asp as they are isomeric, resulting in the same b- and y-fragments derived from CID. Therefore, their differentiation often leveraged chromatographic retention times.3 Since the elution order of Asp and isoAsp can change depending on the chromatographic conditions6 , this indirect approach lacks reliability.
In contrast, alternative fragmentation techniques can produce cand z-fragments indicative of isoAsp. After an electron is attached to an isoAsp, electron rearrangements can lead to a bond breakage between the α-carbon and the methylene group in the peptide backbone, producing c+57 and z-57 fragments (Figure 3).7 These ions are not present when fragmenting Asp due to the lack of a methylene group in the peptide backbone. Although alternative fragmentation technology has been used for this type of identification in the past, the application suffered from overall low sensitivity, with the usage of nano-flow regimes and selective reaction monitoring instead of automated DDA workflows.4,7 This largely limited the adoption in the biopharmaceutical industry.
With the SCIEX ZenoTOF 7600 system, an alternative fragmentation is introduced, enabling scientists to get an in-depth picture of their samples by using analytical flow liquid chromatography (LC) separation in combination with a fast scanning DDA method and automated processing using Protein Metrics Inc. software. This breakthrough technology realizes the dream of high-throughput fragmentation in the biopharmaceutical industry capable of answering complex questions in a routine manner.
Identification and localization of deamidations
The study focused on the characterization of a commercialized mAb: adalimumab. Multiple potential deamidation sites are present in the sequence of adalimumab and previous high pH stress studies on the intact protein suggest that several sites are highly accessible for deamidation events and isomerization of Asp.8 However, the intact mass analysis approach could not reveal the exact localization of these events.
Here, a DDA approach in combination with the patented Zeno EAD was chosen. With this approach, routine peptide mapping analyses can be performed, while the EAD enables advanced characterization in the same, single analysis. Furthermore, the detection of fragment ions and thus the correct identification of low abundant deamidations or Asp isomerizations is enhanced by the Zeno EAD. This approach allowed for the straight-forward data interpretation using Byos software (Protein Metrics Inc.).
Deamidation sites could be accurately identified as shown for the peptide NSLYLQMNSLR (Figure 4). The high MS/MS sequence coverage of >80% with excellent signal-to-noise level provided great confidence in the assignment of both, non-modified and the deamidated species. The modification site could be clearly localized by the mass shift of z4 and c8 (and greater) of 0.98 amu compared to the unmodified peptide.
Differentiation of Asp and isoAsp
The identification and localization of deamidations and associated isomerizations can be further complicated if multiple potential sites are present in the same peptide: Peptide SGTASVVCLLNNFYPR contains two potential deamidation sites (asparagine; N) right next to each other. The main form without any modification and three deamidated forms were found (Figure 5). Interestingly, two modified forms eluted before the main peak and one thereafter (in Figure 5A and 5B). The Zeno EAD spectra revealed that in two cases the site of modification was found to be the N-11, clearly identified by a mass shift of z6, but not z5 ions, compared to the unmodified peptide. This excludes the N12 position as the site of modification. The difference of the two modified peptides in Figure 5A and 5B is localized to the side chain of the asparagine, which results in the presence or absence of c10+57 and the z6-57. For the peak eluting prior to the main species, the diagnostic ions for isoAsp (c10+57 and z6- 57) were found, confirming that the peptide eluting first contains the isoAsp (Figure 5A). In addition, the deamidated version of the peptide at position N-12 was found to be eluting prior to the main species (Figure 5C). Again, this peptide and its modification was clearly identified by characteristic ions. In this case, the elution time is consistent with previous predictions that isoAsp has an earlier elution time compared to the non-modified peptide, and the deamidated version.1 However, the elution of isoaspartic acid does not always follow this rule.3 The peptide VVSVLTVLHQDWLNGK in adalimumab shows an example for a deamidation and associated isoAsp formation for which the mentioned elution order is partly reversed (Figure 6). Two deamidation peaks were observed for this peptide, which elute after the non-deamidated peak (Figure 6A and 6B). The automated assignment of the EAD spectra proves that N-14 represents the modification site for both peaks as z-ions greater than z-4 all showed a 0.98 amu mass shift (Figure 6A and 6B). However, the diagnostic ions z3-57 and c13+57 were only detected in the peak eluting right after the main form, confirming isoAsp formation (Figure 5A).
Unambiguous identification, localization and differentiation of deamidations and isoAsp formations in one single DDA run with automatized data interpretation was achieved with the SCIEX ZenoTOF 7600 system using Zeno EAD. This is an example of how distinguishing isomers previously thought a challenge by LC-MS/MS can be simplified in a reproducible manner by EAD in combination with the Zeno trap (Zeno EAD). This strategy can also be applied for further PTM analysis as well as to the differentiation of leucine and isoleucine for full confidence in the correct sequence of biopharmaceuticals in development.9,10
 Click to enlarge
Click to enlarge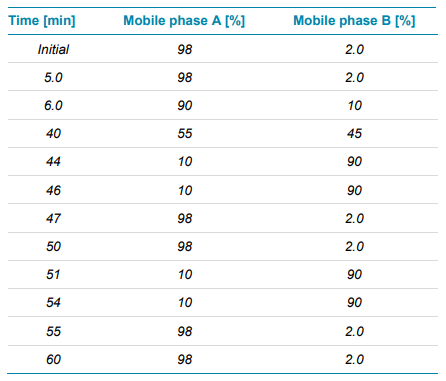 Click to enlarge
Click to enlarge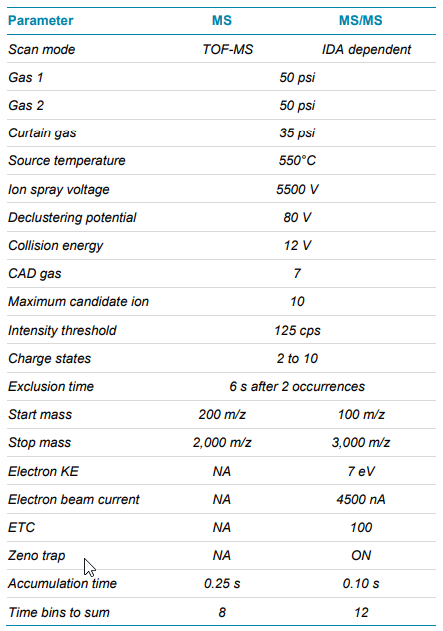 Click to enlarge
Click to enlarge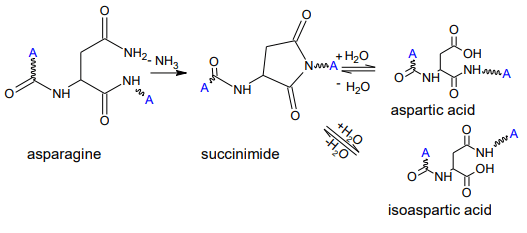 Click to enlarge
Click to enlarge Click to enlarge
Click to enlarge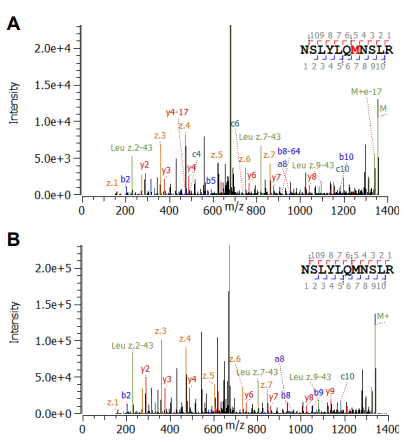 Click to enlarge
Click to enlarge Click to enlarge
Click to enlarge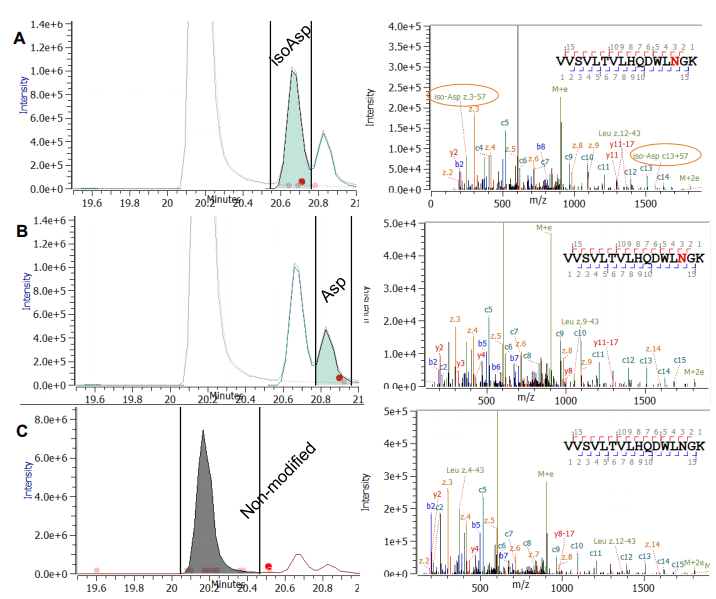 Click to enlarge
Click to enlarge