Simple, fast and robust method for plasmid purity testing and degradation monitoring
Capillary Electrophoresis-Laser Induced Fluorescence Detection (CE-LIF)
Jane Luo, Tingting Li, and Mukesh Malik
SCIEX, Brea, California
Abstract
This study demonstrates the sensitive quantitative analysis of 5 kb and 7 kb plasmid molecular isoforms with the PA 800 Plus equipped with laser-induced fluorescence in a highly automated workflow. An important aspect of the workflow involves dilution of the eCAP dsDNA 1000 gel with 1X TBE buffer in order to provide highly resolved plasmid isoforms in a short period of time. In less than 12 minutes, baseline separation of supercoiled, linear and open circular plasmid isoforms is achieved. The method is capable of monitoring low-level plasmid degradants with 0.23% RSD for migration time and 0.61% RSD for % area. Analysis of plasmids larger than 7 kb or significantly smaller than 5 kb can be achieved with more or less dilution of the dsDNA 1000 gel. Compared with slab gel methods that use ethidium bromide, the LlFluor EnhanCE fluorescent stain used in this method is safer. Additionally, less waste is generated, sample handling is minimized, sample loading is automated, greater sensitivity and reproducibility are observed and more accurate quantification is achieved.

Introduction
Plasmid DNA is often used as the gene delivery vehicle for vaccinations and gene-therapy products. It is also an important intermediate in making Adeno-associated viruses and lentiviruses. The Food and Drug Administration (FDA) guidelines recommend manufacturers include a specification of > 80% supercoiled plasmid content for bulk release (Ref 1). Capillary electrophoresis with laser-induced fluorescence detection (CELIF) provides a rapid, sensitive, reproducible and automated method for the quantitative analysis of plasmid DNA isoforms. Advantages of CE-LIF over slab-gel electrophoresis include rapid analysis time, minimal sample handling, simpler set up and preparation time, greater sensitivity and reproducibility, more accurate quantitation, automated sample loading, and reduced waste generation. This technical note describes a CE-LIF method for plasmid analysis. In this method, the supercoiled, linearized and open circular plasmid isoforms for 5 kilobase and 7 kilobase plasmids can be reproducibly separated with baseline resolution in less than 15 minutes.
Key Features
- Complete workflow solution for plasmid analysis including hardware, software and reagent kits
- Baseline resolution of supercoiled, linear and open circular plasmid isoforms
- Sample separation takes less than 12 minutes
- Increased throughput for analysis via automated sample separation and data analysis
- Robust, good repeatability quantitative assay with RSD of 0.23% for migration time and 0.61% for % area
- Capable of monitoring low-level plasmid degradants
- Less waste generation than slab gel method
- LIFluor EnhanCE stain in this method is safer than ethidium bromide used in slab gel method
Figure 2. Confirmation of different isoforms in an untreated 5 kb plasmid preparation (A) using linearized (B) and nicked plasmids (C) as controls. The major peak in the untreated Plasmid Preparation (A) is the supercoiled isoform; the smaller peak is the open circular isoform. In the “Mixed” sample (D), the first peak is the linear isoform, the second peak is supercoiled isoform, and the third one is the open circular isoform.

Materials and Instrument
Materials:
The eCAP dsDNA 1000 kit (PN 477410, Figure 1B), the LIFluor EnhanCE fluorescent stain (PN 477409, Figure 1C), dsDNA 1000 Test Mix (PN: 477414) and LIF Performance Test Mix (PN: 726022) were from SCIEX, Framingham, MA. A 10X concentrated stock solution of Tris Borate EDTA (10x TBE, PN T4323) was from Sigma Aldrich, St. Louis, MO. The 5 kilobase (kb) Plasmid DNA (PN E1541) was from Promega, Madison, Wisconsin. The 7 kb Plasmid DNA was from an internal project at SCIEX, Framingham, MA. The nicking enzyme, Nb.BsmI (PN R0706) and BamHI-HF restriction enzyme (PN R3136) were from New England BioLabs, Boston, Massachusetts.
Instrument and software: A PA 800 Plus Pharmaceutical Analysis System (Figure 1A) equipped with LIF detection and solid-state laser with excitation wavelength at 488 nm and a 520 nm band pass emission filter were from SCIEX, Framingham, MA. Data acquisition and analysis were performed using 32 Karat software V10.
Methods
Gel buffer reconstitution: This step must be done one day before running samples. To rehydrate the gel buffer, 20 ml of 0.2 µm filtered deionized water was added to the gel buffer vial. After the gel buffer vial was securely capped, the vial was placed on a rotator or the gel mixture was gently stirred with a small stirring bar for up to 24 hours or till the dried gel was completely dissolved. The hydrated gel should be good for one month if stored at 4oC. Then, the gel solution was diluted 1:10 with 1X TBE (90 mM Tris, 90 mM Borate, and 2 mM EDTA pH 8.3). The diluted gel was filtered through a 0.45 µm filter. 1x TBE was prepared by a 10x dilution of TBE stock solution with deionized water followed by passing through a 0.2 µm filter. LIFluor EnhanCE stain was added to the gel at a concentration of 5 μl stain in 6 ml gel (Ref 2).
Cartridge Assembly: DNA capillary (PN 477477) was installed per instructions on kit insert (PN 726412) in the dsDNA 1000 kit. The total capillary length was 40.2 cm with 30 cm as the length to the detection window. Since the inner wall of the DNA capillary is coated, the cartridge assembly should be carried out in a timely manner. Excessive exposure to air may damage the inner coating and also cause clogging. The capillary ends must be immersed in liquid (water or buffer) as soon as the cartridge assembly is complete to prevent the coating from drying out.
Sample Preparation: In order to identify different plasmid isoforms, linearized and open-circular plasmids were prepared as controls. To prepare the linearized isoform, a single-cutting restriction endonuclease, BamHI-HF, was used to digest plasmids following manufacturer’s instructions. To obtain the open circular isoform, plasmid preparations were either partially or fully digested with a nicking enzyme according to the manufacturer’s instructions. All plasmid samples were diluted with deionized water to 10 ng/μl for analysis on the PA 800 Plus instrument.
LIF Calibration: To ensure consistent response of LIF detector throughout this study, the LIF detector was calibrated using LIF Calibration Wizard and Performance Test Mix (PN: 726022) following the instructions in LIFluor EnhanCE user’s guide (PN 725824). The target RFU value was set as 62. This calibration was done whenever capillary or LIF detector or the laser was changed. After the calibration was complete and the CCF was accepted, vials of water were placed back to the “home” positions (BI:A1 and BO:A1) so that the capillary ends were placed in water to keep them wet.
Instrument Performance Test (optional): Instrument and gel buffer performance may be tested using dsDNA 1000 Test Mix (PN 477414) and LIFluor EnhanCE stain (PN 477409). Test Mix can be diluted by adding 1 mL of filtered deionized water (> 18MΩ) to the test mix vial. 5 µl LIFluor EnhanCE stain may be added to 6 mL of 1X rehydrated dsDNA 1000 gel buffer. Separation may be carried out following instructions in LIFluor EnhanCE user’s guide (PN 725824).
Preparation of Buffer Trays and Sample Trays: Vial positions for buffer trays are indicated in Figure 3. Each “DDI Water” vials were filled with 1.5 ml double deionized (DDI) water. Waste vials were filled with 1 ml DDI water. “Gel Fill” vials were filled with 1.5 ml gel. “Gel Sep.” vials were filled with 1.2 ml gel. Vial “A6” on the outlet was filled with 1 ml gel. Two sets of water dip vials were set up for minimal sample carryover and better repeatability. Injecting samples against a half-filled vial of gel buffer (BO:A6) facilitated improved precision in peak area. Each plasmid sample was transferred to a Microvial (PN 144709) at 95 to 100 μl. Sample vials were loaded onto the sample inlet tray.
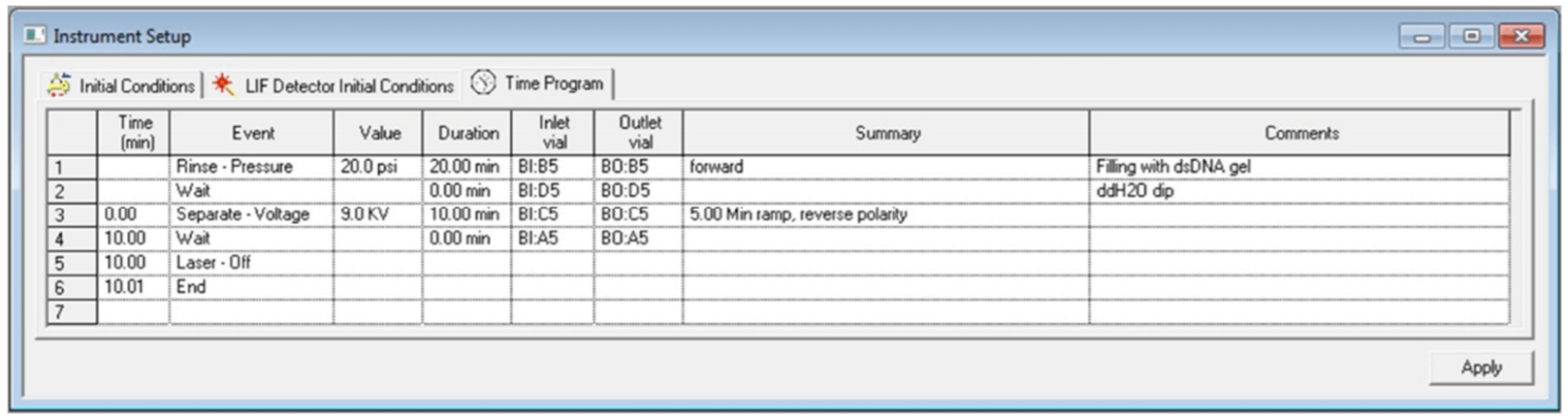
Instrument Set up:
The “Initial Conditions” and “LIF Detector Initial Conditions” were set up as indicated in Figure 4 and Figure 5 respectively. Same set up was used for all three methods: Capillary Conditioning, Separation and Shut Down.
The time program for Capillary Conditioning was illustrated in Figure 6. Buffer vials used for conditioning were purposely positioned in row #6 to avoid moving the capillary tips over buffer vials used for sample separation. Figures 7 and 8 show time programs for plasmid separation and shut down methods.
Figure 3. Schematic set up for Inlet (left) and Outlet (right) buffer trays.
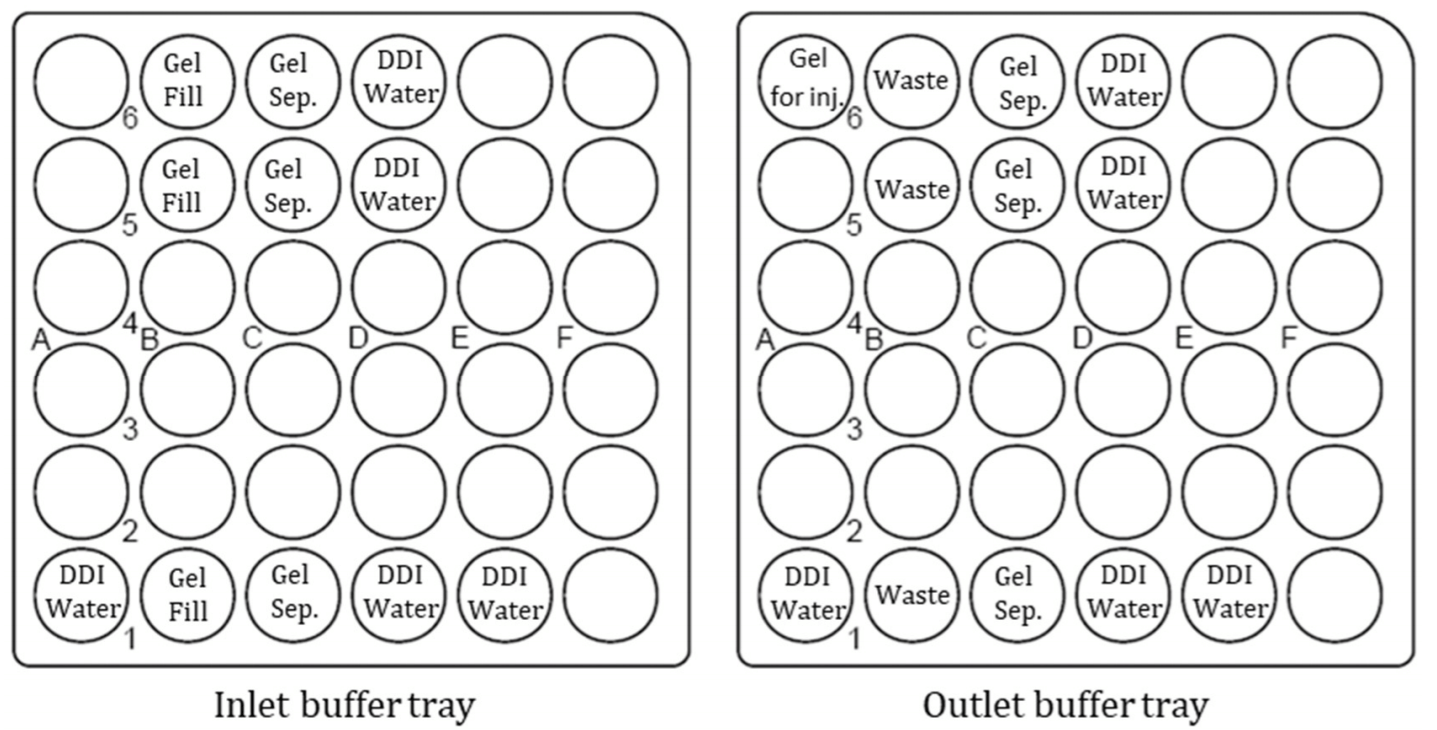
Figure 4. Settings for “Initial Conditions” tab.
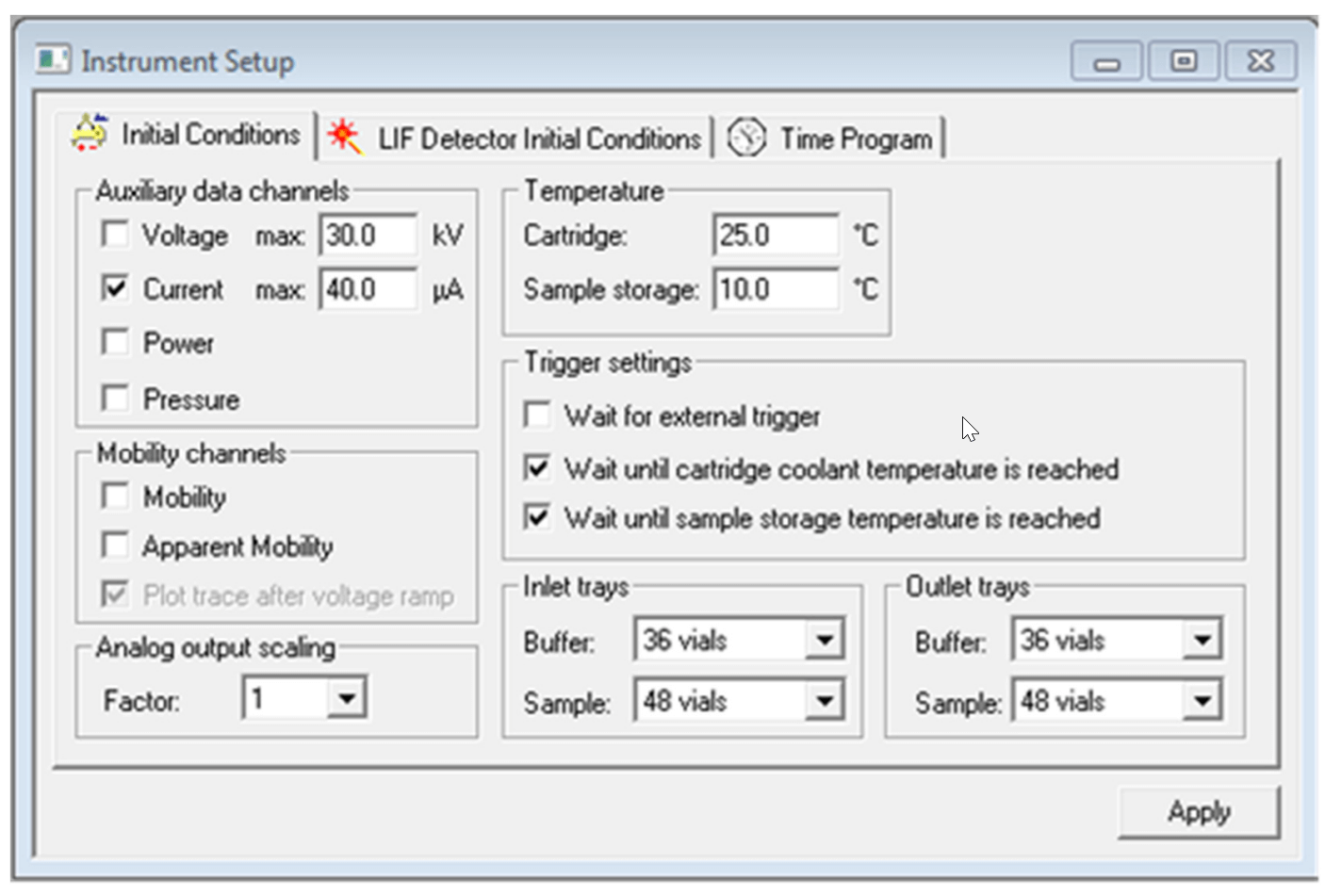
Figure 5. Settings for “LIF Detector Initial Conditions” tab.
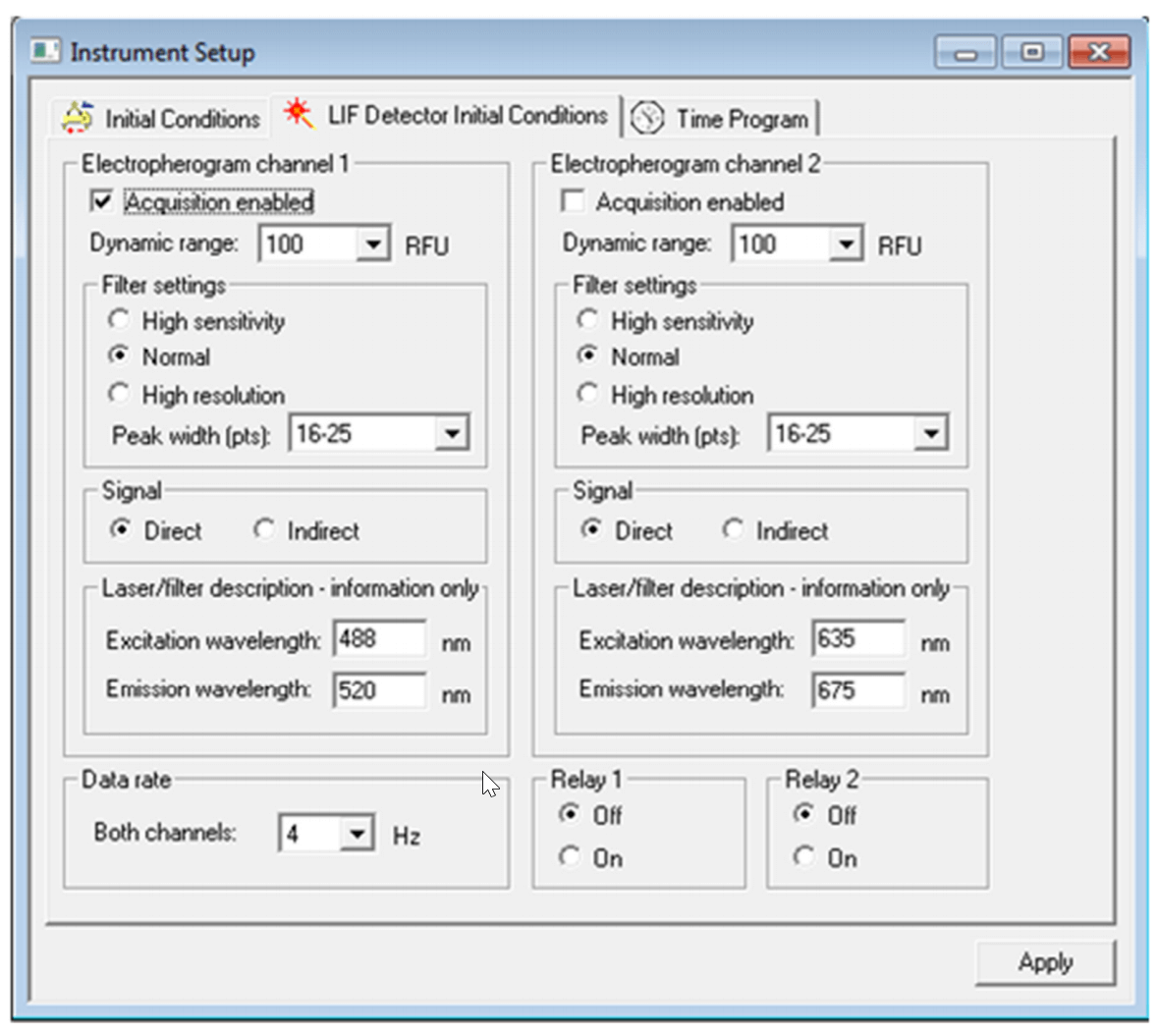
Figure 6. Time Program Settings for the Conditioning Method.
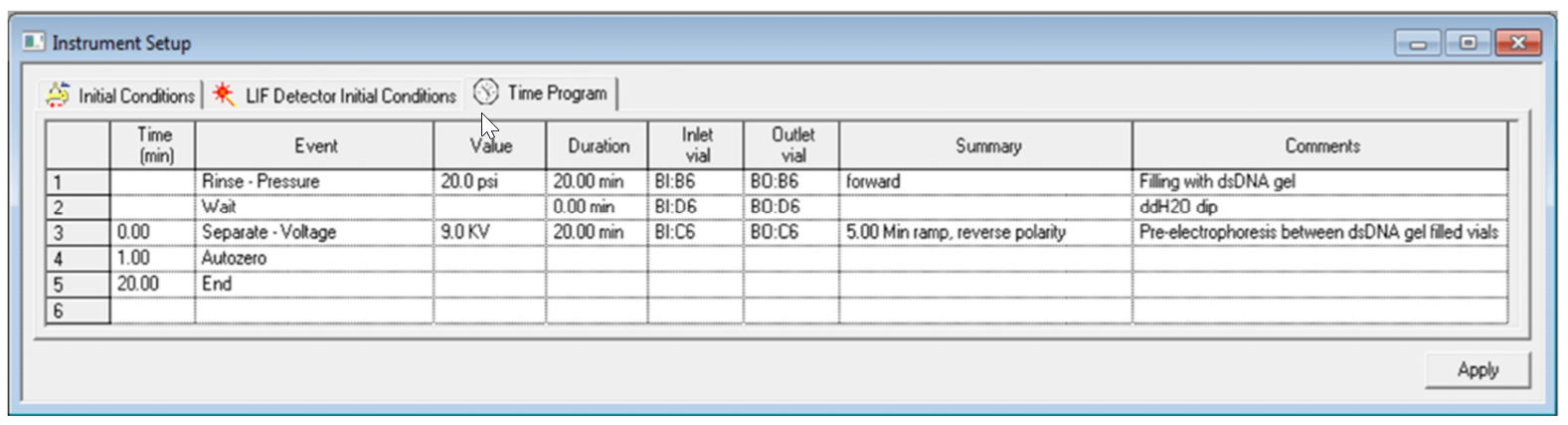
Figure 7. Time Program Settings for the Separation Method.

Figure 8. Time Program Settings for the Shut Down Method.
Results
An optimized method to resolve and quantitate plasmid isoforms via capillary electrophoresis using commercially available eCAP dsDNA gel buffers is described in this technical note. When gel buffer from eCAP dsDNA 1000 kit was used without further dilution, it was found to entrap the supercoiled plasmid DNA, causing long separation times and broad circular plasmid peaks. However, by simply diluting the eCAP dsDNA 1000 gel with 1X TBE buffer, the diluted gel demonstrated the ability to provide highly resolved plasmid isoforms in a short period of time. In the case of the 5 kb test plasmid, the best results were obtained through a simple 1 to 10 dilution of dsDNA 1000 gel buffer in 1X TBE. Because the separation of the plasmid isoforms in eCAP dsDNA gels is achieved by the trapping of the supercoiled and circular isoforms of plasmid, the migration of these isoforms is slowed relative to that of the linearized molecule. This resulted in a migration pattern that is opposite to that typically observed in agarose gels where the super-coiled form precedes the linearized. The method presented here was optimized to work with LIF detection using LIFluor EnhanCE stain.
Analysis of a 5 kb plasmid
Figure 9 shows the results obtained with a 5 kb plasmid preparation. The main isoform is the supercoiled form while the minor isoform is the open circular form. To confirm this peak assignment, a comprehensive experiment was performed where a linearized isoform was prepared by digesting the 5 kb plasmid with a single cutting restriction enzyme, BamHI-HF. In addition, a partial digestion of the plasmid with a nicking enzyme was also carried out to simulate the normal degradation of plasmid. Then, the untreated plasmid preparation, the linearized sample, the partially nicked sample and a mixture of the three were separated on the PA 800 Plus and compared to each other. In Figure 2, results showed that the linearized form migrated in front of the supercoiled form. The partially nicked sample contained both the supercoiled form and the open circular form as well as a trace amount of the linearized form. As expected, the amount of nicked or open circular form increased while the supercoiled form decreased in the partially nicked sample relative to those in the untreated plasmid preparation. These results indicated that this plasmid separation method is capable of monitoring plasmid degradation and also provide confirmatory information for peak assignments. In the mixed sample, all three isoforms were baseline resolved.
There is a small peak between the supercoiled form and the open circular form which could be the supercoiled dimer form. Further experiments are needed to confirm this hypothesis.
Figure 9. Separation of typical plasmid isoforms in a 5 kb plasmid preparation.

Figure 10. Separation of plasmid isoforms in a 7 kb plasmid preparation.
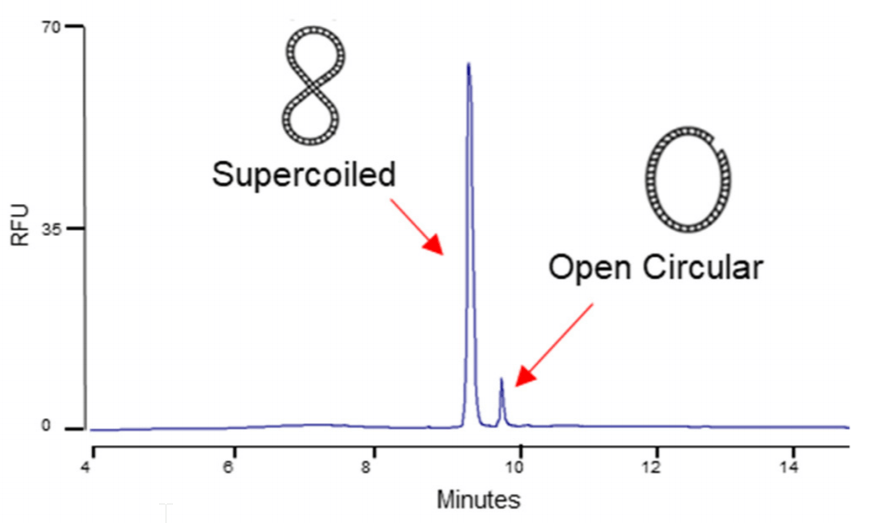
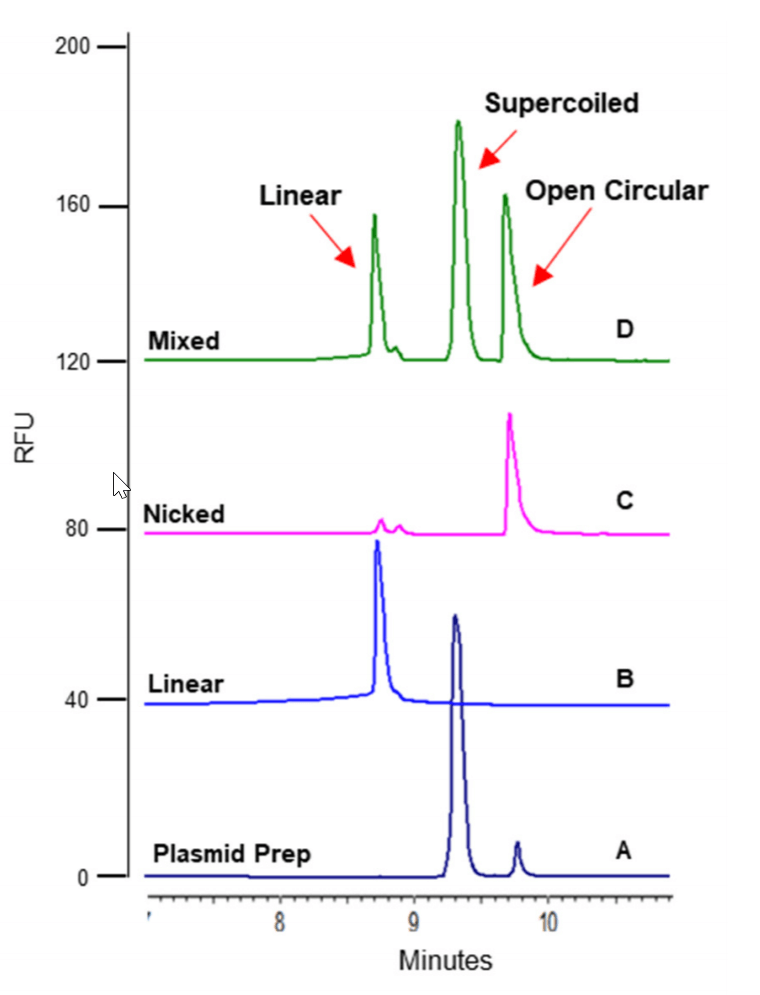
Figure 11. Confirmation of different isoforms in an untreated 7 kb plasmid preparation (A) using linearized (B) and nicked plasmids (C) as controls. The major peak in the untreated Plasmid Preparation (A) is the supercoiled isoform; the smaller peak is the open circular isoform.
Analysis of a 7 kb plasmid
The plasmid analysis method using dsDNA 1000 gel, coated DNA capillary and LIFluor EnhanCE stain was further evaluated using a 7 kb plasmid. In Figure 10, results show a baseline resolution of the supercoiled form and the open circular form. The amount of open circular form was very low. There was no other isoform present, indicating that this is a high quality plasmid preparation. Further experiments were carried out to ensure that the small open circular peak was the nicked isoform. A linearized isoform was prepared by digesting this 7 kb plasmid with a single cutting restriction enzyme, BamHI-HF. In addition, a complete digestion of the plasmid with a nicking enzyme was also carried out. At the end of the digestion with the nicking enzyme, the reaction mixture was also heated at 80oC for 20 min to inactivate the enzyme. Then, the untreated plasmid preparation, the linearized sample, the fully nicked sample and a mixture of the three were separated on the PA 800 Plus and compared to each other. In Figure 11, results shows the small peak in the untreated plasmid preparation was indeed the open circular form. In the fully nicked plasmid sample, there was no supercoiled isoform present. In addition, there was a small amount of linear form present possibly due to further degradation during the heat inactivation of the nicking enzyme. Once again, the results in this experiment indicates that this plasmid analysis method provides accurate assessment of the isoforms in plasmid preparation and is also capable of monitoring plasmid degradation.
Method robustness and repeatability
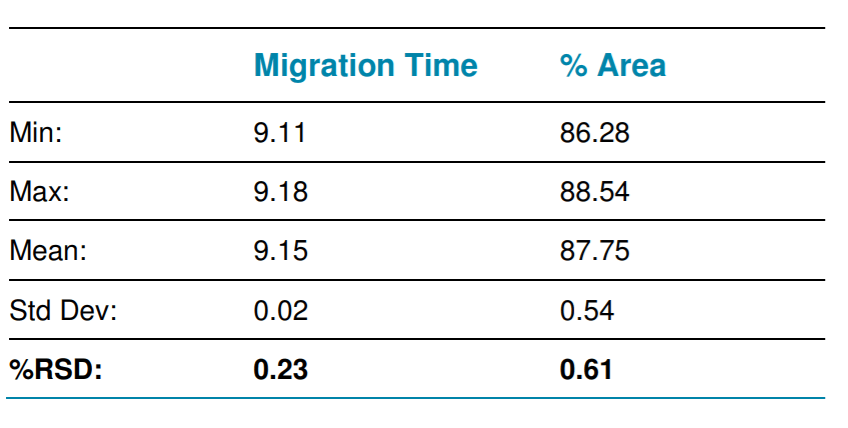
The robustness and repeatability of this method was evaluated by performing 18 runs of the same untreated 5 kb plasmid sample. As shown in Figure 12, the peak profiles were very consistent between 18 runs. Table 1 shows the values of % RSD for migration time and % area for the supercoiled isoform obtained with the untreated 5 kb plasmid were 0.23% and 0.61% respectively.
Figure 12. Overlay of 18 runs of untreated 5 kb plasmid preparation.
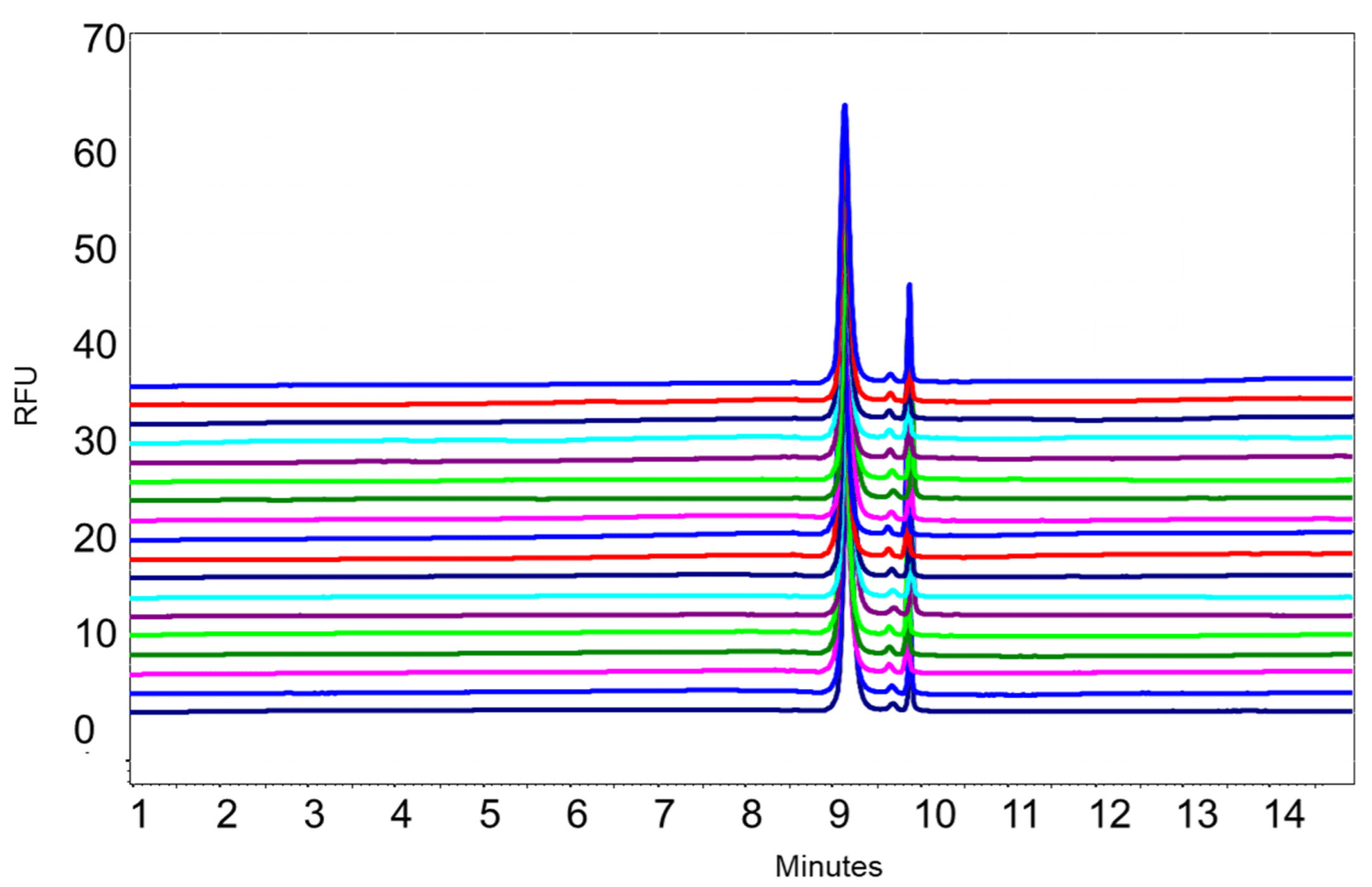
Table 1. Calculation of % RSD for migration time and % area for the supercoiled isoform for 18 runs obtained with the 5 kb untreated plasmid preparation.
Discussion
This plasmid analysis method was initially optimized for the 5 kb plasmid. It also works for the 7 kb plasmid. The resolution of plasmid isoforms for the 5 kb plasmid (Figure 9) was slightly better than that for the 7 kb plasmid (Figure 10). Users may need to further optimize this method by further diluting the dsDNA 1000 gel for analysis of plasmids with sizes larger than 7 kb. Similarly, less dilution of the gel may be needed for analysis of plasmids with sizes significantly smaller than 5 kb. No matter what sizes of plasmid DNA need to be analyzed, preparing linearized and nicked plasmid isoforms as controls is a great strategy in evaluating the accuracy of the plasmid analysis method, as demonstrated in this technical note.
Conclusions
- Fast analysis of plasmid samples with separation time within 12 minutes
- Baseline resolution of supercoiled, linear and open circular isoforms of a 5 kb and a 7 kb plasmid
- The dsDNA 1000 kit and LIFluor EnhanCE stain are commercially available kits for users thus streamlining the workflow
- Automated sample separation and data analysis provide rapid, reproducible and quantitative results with RSD of 0.23% for migration time and 0.61% for % area
References
- FDA. Points to consider on plasmid DNA vaccines for preventive infectious diseases. 1996. Docket no. 96N0400.
- Boardman C. and Dewald H. Plasmid purity and heterogeneity analysis by capillary electrophoresis. Poster at MSB 2010.