Application of EPA method 544 for the analysis of microcystins and nodularin in drinking water
Sujata Rajan, 1 Sashank Pillai1 and Craig M. Butt2
1 SCIEX, India; 2 SCIEX, USA
Abstract
This technical note describes the analysis of 7 microcystins and nodularin in drinking water following United States EPA Method 544.1 The minimum reporting levels (MRL) in the fourth Unregulated Contaminant Monitoring Rule (UCMR4) were confirmed in the initial demonstration of capability (IDC) experiments using the QTRAP 4500 system (Figures 1 and 3). The MRLs met the predicted interval of results (PIR) criteria of 50%–150% recovery and ranged from 5 to 20 ng/L. In addition, the IDC experiments included the assessment of method blanks (<33% MRL) and the determination of accuracy (70–130%) and precision (%CV <20%) in laboratory fortified blanks (LFBs). The use of 250 mL of water samples reduces sample preparation time compared to the traditional 1 L sample volume.
Introduction
Microcystins are toxic cyclic heptapeptides produced by cyanobacteria that occur in natural waters during harmful algal blooms (HABs).2 Microcystins are potent liver toxins and present health risks to humans and animals. As such, drinking water guidelines and health advisory levels have been established at sub- to low-µg/L levels across the world. Sensitive, accurate and precise analytical methods are needed to ensure the protection of human health and the environment.
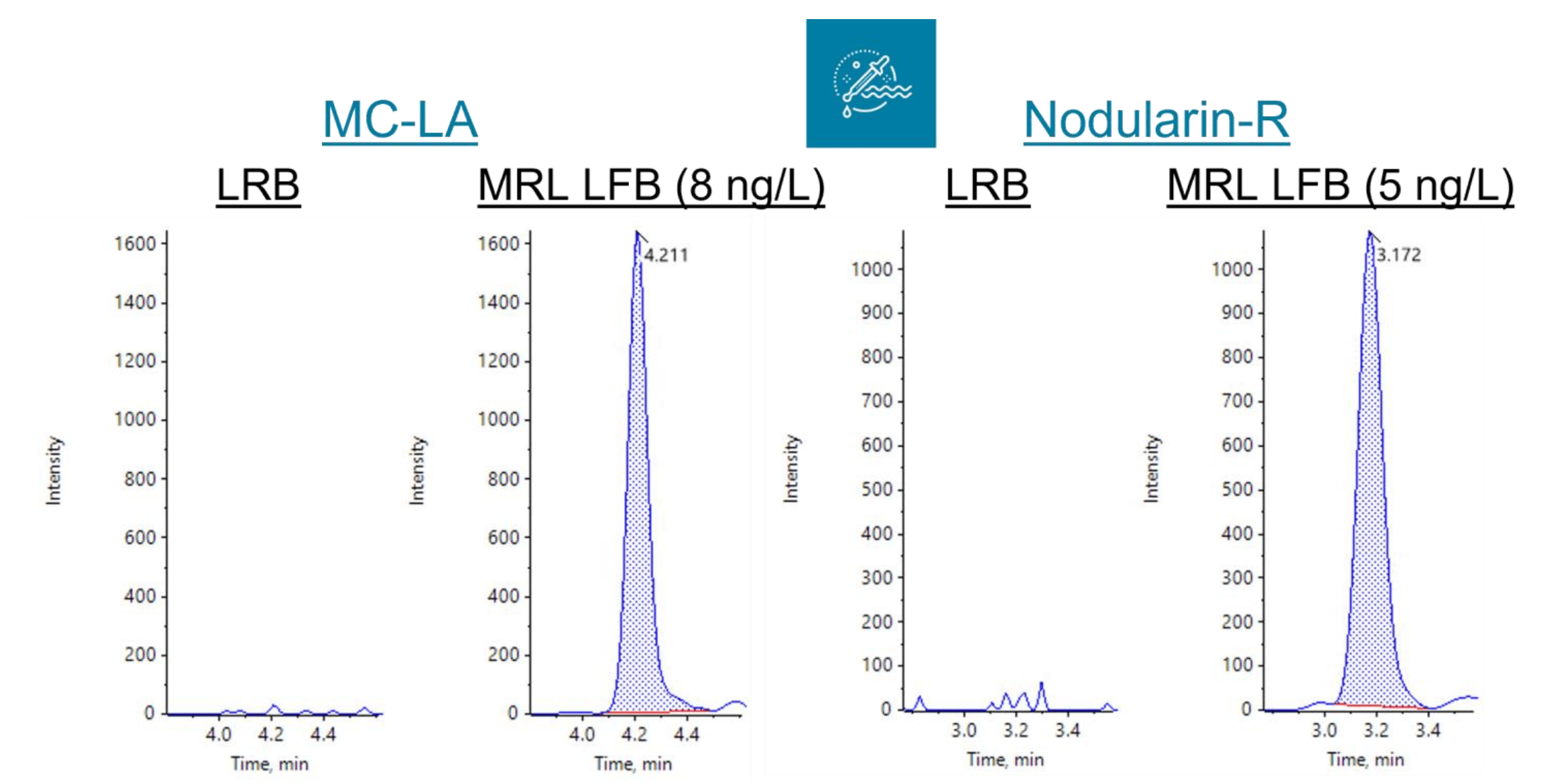
Figure 1. Extracted ion chromatograms (XICs) of laboratory regent blank (LRB) and MRL laboratory fortified blank (LFB) samples for MC-LA (left panels) and nodularin-R (right panels).
Key features for the analysis of EPA Method 544 using the QTRAP 4500 system
- MRL confirmation at UCMR4 reporting levels. Analysis of 7 replicate samples showed PIR limits between 50% and 150% for all analytes, confirming MRLs ranging from 5 to 20 ng/L (0.005 to 0.02 µg/L)
- Accuracy and precision surpassed IDC criteria. LFB spikes (n=4) at 5x the UCMR4 MRL met the EPA Method 544 requirements of ±30% and <30% for accuracy and precision, respectively
- Low ng/mL sensitivity for calibration standards on the QTRAP 4500 system. Solvent-based calibration standard limits of quantitation (LOQ) ranged from 0.5 to 2.5 ng/mL
- Fast 10 min chromatographic gradient. Reduced gradient runtime obtained good chromatographic separation and analyte peak shape
Methods
Standards. Ethylated D5 microcystin-LR (MC-LR-C2D5, 98%), was purchased from Cambridge Isotope Laboratories and used as the surrogate standard. Individual microcystin (RR, LR, YR, LF, LW, LY, LA) and nodularin R standard solutions were purchased from CIFGA. The surrogate standard spiking stock and calibration standards were prepared in 90:10 (v/v), methanol/water. Calibration standard concentrations ranged from 0.05 to 100 ng/mL and contained 30 ng/mL of the surrogate standard.
IDC experiments. IDC experiments followed the procedures described in section 9.2 of the EPA Method 544 document and are briefly described below. Individual sections of the document are highlighted below. Spiking concentrations were chosen to represent the UCMR4 minimum reporting level concentrations. Note that microcystin-LW was not included in the UCMR4 program.
Initial demonstration of low system background (EPA Method 544, section 9.2.1). The laboratory reagent blank (LRB, n=1) consisted of 250 mL of blank laboratory water that was spiked with the surrogate standard and carried through the sample preparation procedure (EPA Method 544, section 11.2).
Initial demonstration of precision and accuracy (EPA Method 544, sections 9.2.2 and 9.2.3). The LFB samples (n=4) consisted of 250 mL of blank laboratory water spiked with the microcystins and nodularin at 5x the UCMR4 levels. As permitted by EPA Method 544, section 8.1.1, the sample volume was reduced to 250 mL and the volumes for solvent rinsing and washing were adjusted accordingly. The final reconstitution volume was 500 μL. Sample preparation details are thoroughly outlined in EPA Method 544, section 11. Briefly, the 250 mL water sample was fortified with the surrogate standard and filtered using an Isopore hydrophilic polycarbonate membrane filter (0.4 μm pore size, 47 mm diameter). The filter was incubated with 80:20 (v/v), methanol/water for 1 hr at -20°C. The filtrate and incubation solution were combined and passed through a solid-phase extraction cartridge (Waters Oasis HLB, 6 cc, 150 mg), eluted with 90:10 (v/v), methanol/water, evaporated to dryness and reconstituted with 90:10 (v/v), methanol/water.
MRL confirmation (EPA Method 544, section 9.2.4) at the UCMR4 levels. To confirm the MRL, 7 LFB samples were spiked at the UCMR4 level. Sample preparation methods followed those for the initial demonstration of precision and accuracy experiments.
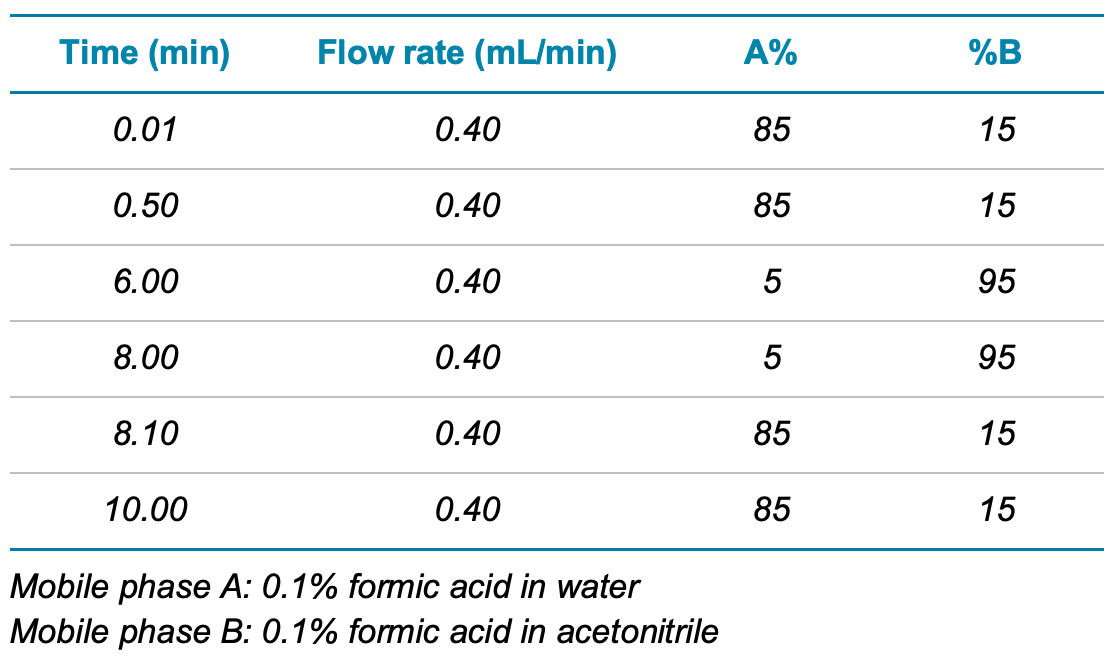
Chromatography. An ExionLC AD system was used with a Phenomenex Kinetex C18 column (2.6 µm, 100 Å, 100 mm x 2.1 mm, P/N: 00D-4462-AN) for the chromatographic separation. The gradient conditions used are shown in Table 1. A flow rate of 0.40 mL/min and a 10 µL injection volume were used. The autosampler and the column oven temperature were set to 8°C and 40°C, respectively.
Table 1. Chromatographic gradient used for the analysis of EPA Method 544.
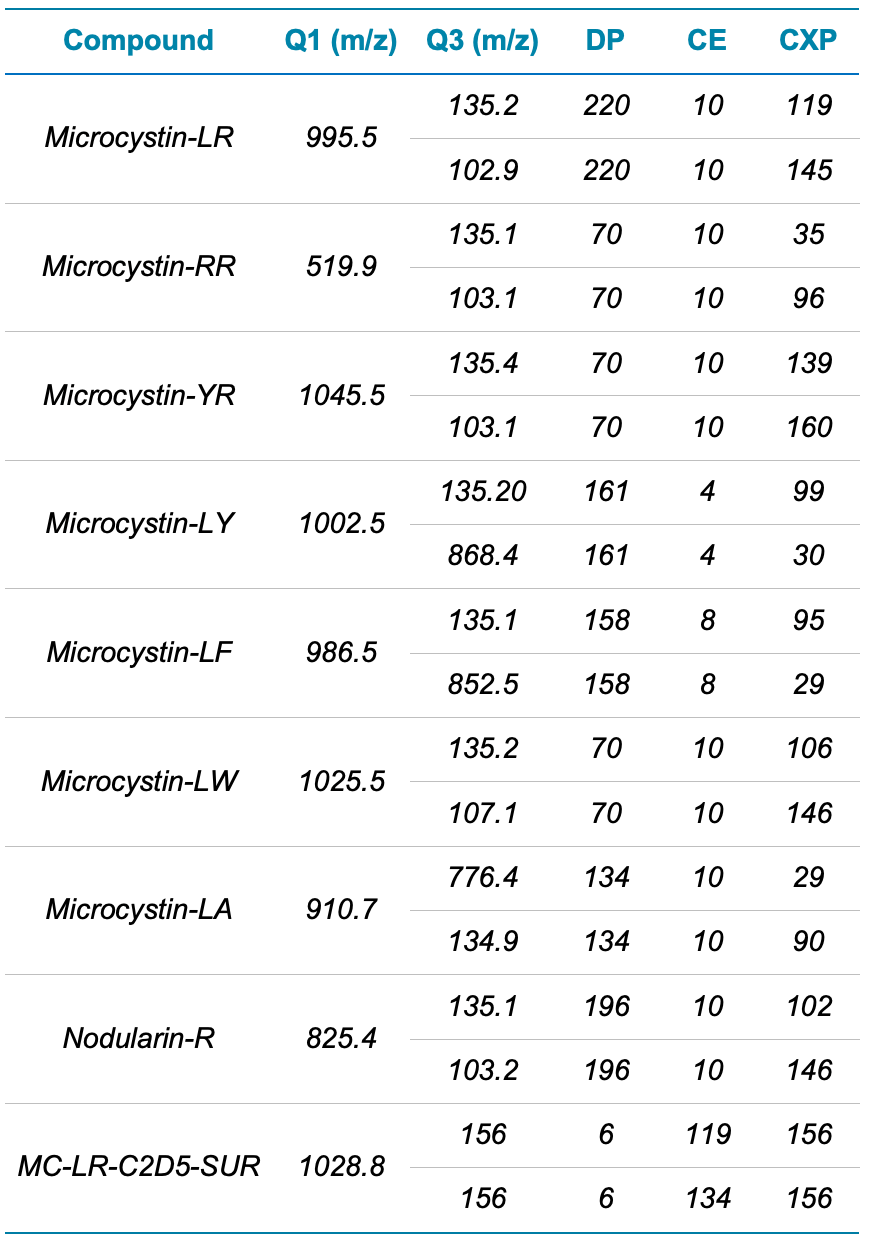
Mass spectrometry. The QTRAP 4500 system was operated in multiple reaction monitoring (MRM) mode with electrospray ionization in positive ion mode. Two selected MRM transitions were monitored for analyte quantitation and confirmation based on the ion ratio. Optimized source and compound-specific parameters are presented in Tables 2 and 3, respectively.
Data processing: All data were processed using SCIEX OS software, version 2.1.6. Analyte responses were normalized to the surrogate standard response. The calibration curve used linear regression and 1/x weighting.
Table 2. Source and gas parameters for the analysis of microcystins and nodularin on the QTRAP 4500 system.
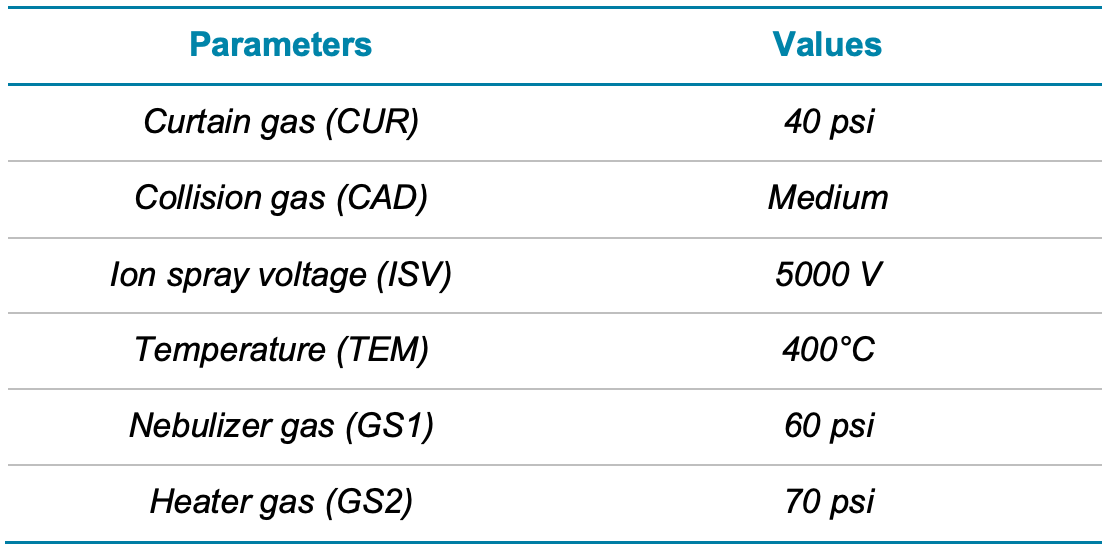
Table 3. Compound-dependent parameters for the analysis of microcystins and nodularin on the QTRAP 4500 system. For each analyte, the top Q3 ion represents the quantifier fragment ion and bottom Q3 ion represents the qualifier fragment ion.
LC optimization for microcystin analysis
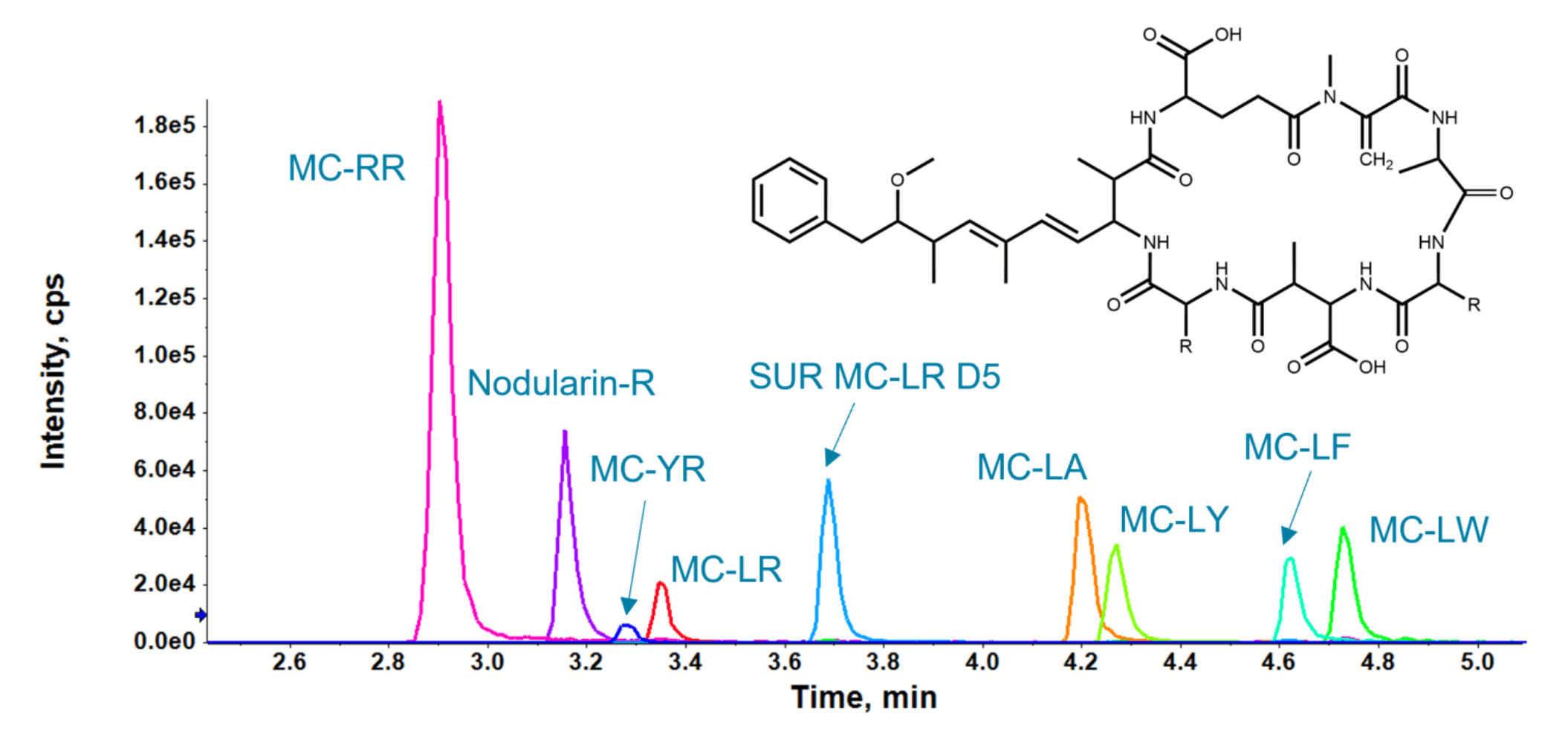
Chromatographic conditions were optimized to achieve a relatively fast runtime of 10 min, while separating potentially interfering, coeluting compounds. Various LC columns, mobile phases and gradients were screened to develop the final method. The 10 min gradient resulted in baseline separation of the 8 cyanotoxin analytes and data quality was confirmed in the MRL experiments. As a result, Figure 2 shows good peak symmetry with optimal datapoints across the quantifier ion peaks at the UCMR4 MRL for all analytes.
Figure 2. XICs of the target microcystins and nodularin-R in the 50 ng/mL solvent calibration standard. The general microcystin structure is shown in the top right.
Sensitivity, accuracy, precision and linearity of solvent calibration standards
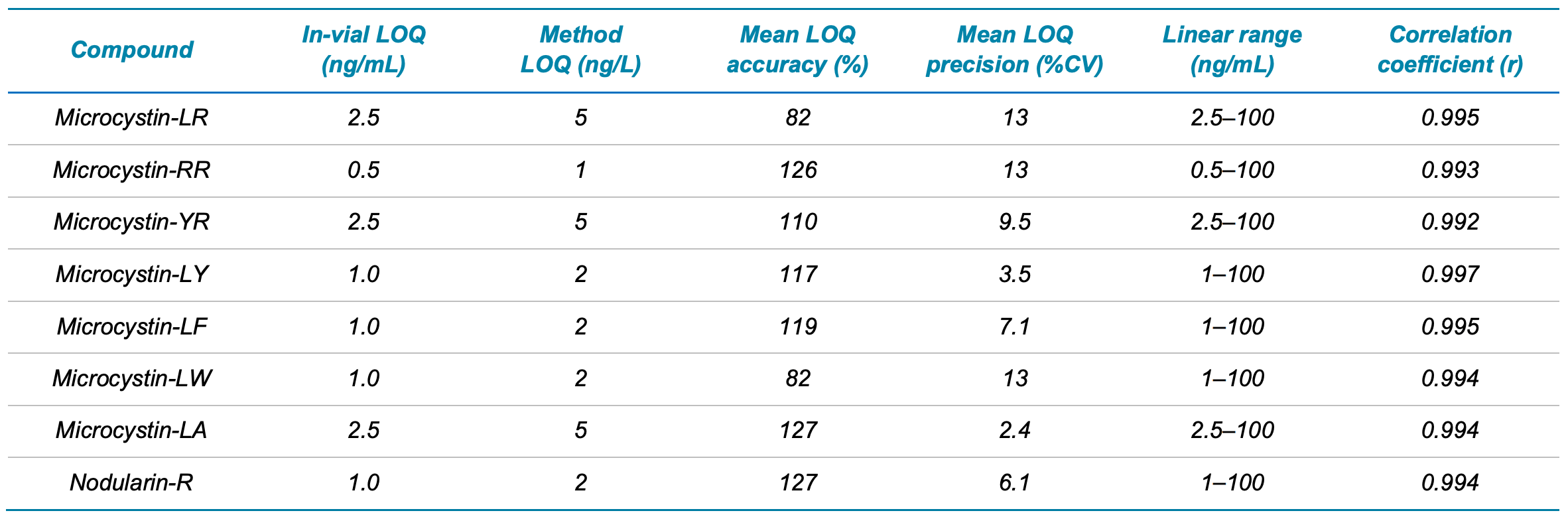
Performance of the QTRAP 4500 system for microcystin and nodularin-R analysis was investigated through triplicate injections of the solvent-based calibration standards. LOQs ranged from 0.5 to 2.5 ng/mL (Table 4), demonstrating good sensitivity of the QTRAP 4500 system. The equivalent method in-sample LOQs ranged from 1 to 5 ng/L and were back-calculated from the in-vial LOQ values based on the 500x SPE concentration factor. Selection of the LOQ was based on the concentration level achieving average accuracy ±30%, precision %CV <15% for the quantifier ion, ion ratio tolerance ±30%, signal-to-noise ratio (S/N) >10 and r value >0.99.
Mean accuracy was between 82% and 127% and the %CV was between 2.4% and 13% at the LOQ. The calibration curve linear range showed r values >0.99 for all analytes.
Table 4. In-vial LOQ (ng/mL), method LOQ (ng/L), mean accuracy (%) and precision (%CV) at the LOQ, calibration curve linear range (ng/mL) and correlation coefficient (r) for the 7 microcystins and nodularin-R analyzed by EPA Method 544. The method LOQ was determined by backcalculating the experimentally determined in-vial LOQ using the 500-fold SPE concentration factor.
IDC experiments
Section 9.2 of the EPA Method 544 document outlines the IDC experiments necessary for a laboratory to demonstrate analytical proficiency. 1 Experiments include the demonstration of low system background (9.2.1), demonstration of precision (9.2.2) and accuracy (9.2.3) and MRL confirmation (9.2.4). Experiments were performed to achieve these criteria, indicating the method performance.
Demonstration of low system background. The extracted LRBs did not exhibit any analyte peaks (n=3) except for MC-LR (Figures 1 and 3). The MC-LR peak originated from the mass-labelled surrogate standard. Overall, these results demonstrate negligible contamination during the sample preparation and instrumental analysis. The criteria that the LRBs must be <1/3 of the MRL concentration were met, including for the MC-LR sample that exhibited minor LRB contamination. Further, none of the analytes were detected in the diluent blanks.
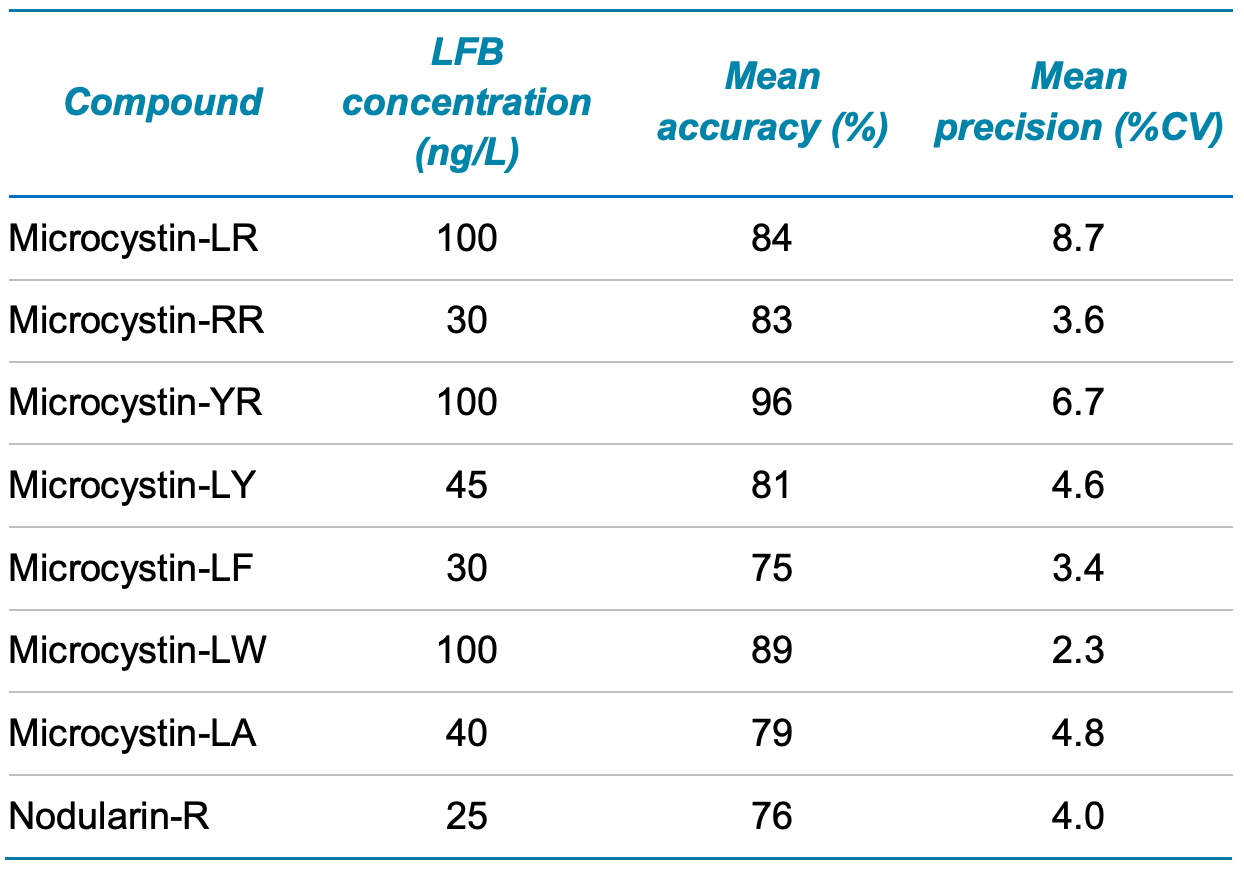
Demonstration of precision and accuracy. LFBs were spiked at 5x the UCMR4 (n=4), representing the mid-point of the calibration curve, and processed through the sample preparation and analysis procedure. The mean LFB accuracy ranged from 75% to 96%. The mean %CV for LFB precision ranged from 2.3% to 8.7%. The accuracy and precision values observed were within the acceptable criteria of ±30% and <30%, respectively (Table 5).
Table 5. Initial demonstration of accuracy and precision. LFBs were spiked at 5x the UCMR4 MRL level (n=4).
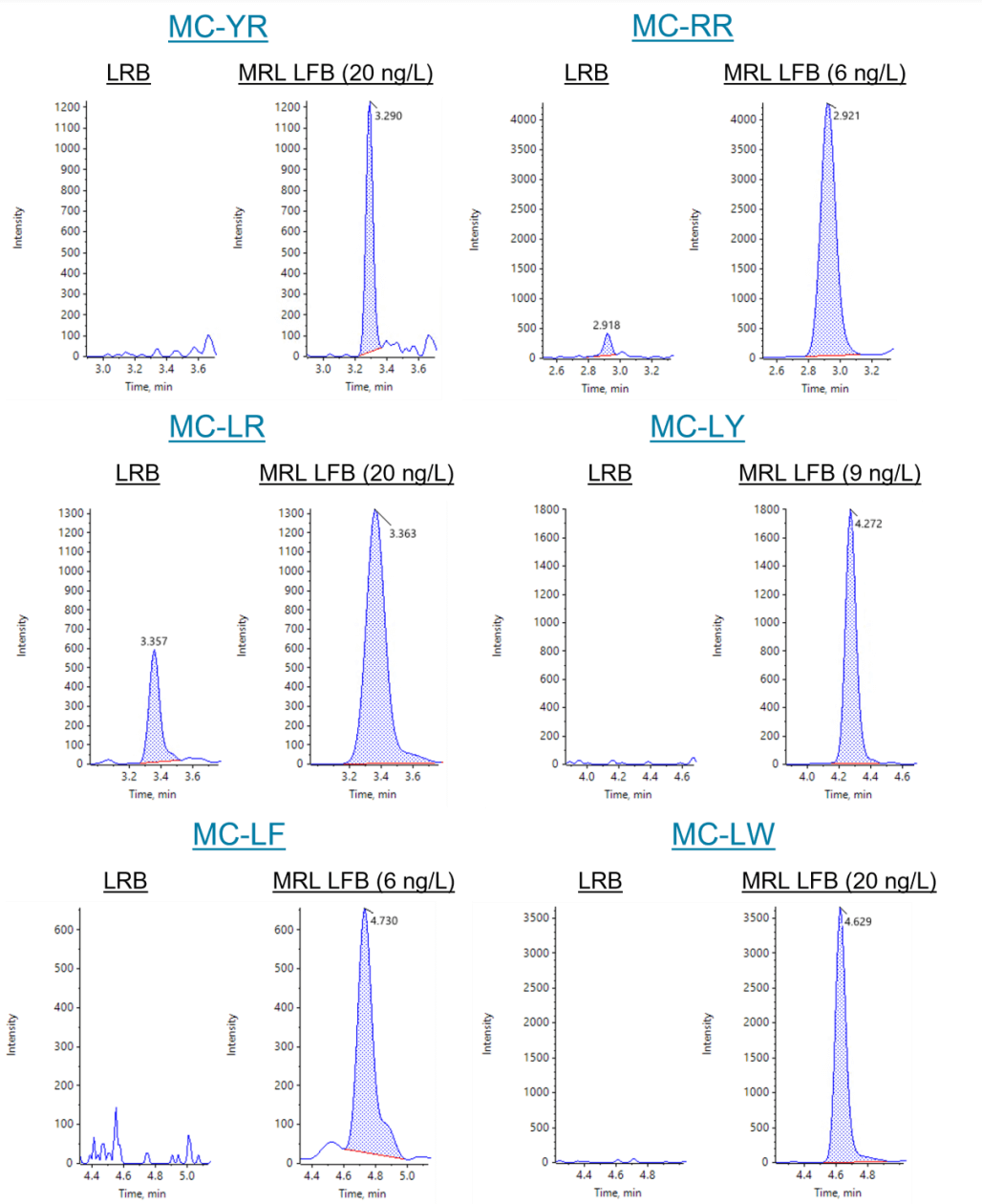
Minimum reporting level confirmation at UCMR4 targets. The UCMR4 reports levels were set as the targeted MRL concentrations for this study (Table 6). To confirm these MRLs, a set of LFBs (n=7) was spiked at the UCRM4 reporting levels and processed through the sample preparation and analysis procedures. MRLs were considered verified if the calculated PIR limits were within 50%–150%. The PIR limits are a measure of recovery accuracy and precision.
Mean recoveries ranged from 88%–117% with %CV ranging from 3.2%–11%. XICs for the LRB and MRL LFB samples are shown in Figures 1 and 3. The mean measured recovery percentage and standard deviation (Table 5) were used to calculate the PIR using the following equation:
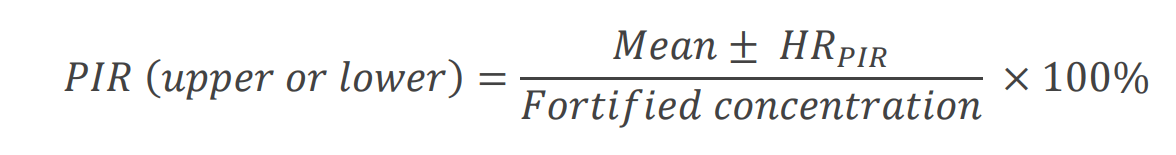 Click to enlarge
Click to enlargeWhere HRPIR represents the half range for the PIR and was calculated as,
𝐻𝑅𝑃𝐼𝑅 = 3.963𝑠
𝐻𝑅𝑃𝐼𝑅 = Half range for the PIR
𝑠 = The standard deviation of replicate analyses
3.963 = Constant value for 7 replicates
The calculated lower PIR values ranged between 53% and 102% and the upper PIR values ranged between 112% and 140%. Therefore, the MRLs were verified at low ng/L levels, achieving the UCMR4 target reporting levels for the analysis of microcystins and nodularin in drinking water. These results demonstrate the ability of the QTRAP 4500 system to analyze samples following EPA Method 544 with sensitivity, accuracy and precision.
Table 6. Confirmation of the MRL at UCMR4 target concentrations. LFB samples (n=7) were spiked at the UCMR4 MRL target concentration and processed through the EPA Method 544 sample preparation and LC-MS/MS procedure. Calculated PIR limits were within the 50%–150% criteria, verifying the MRL concentration.
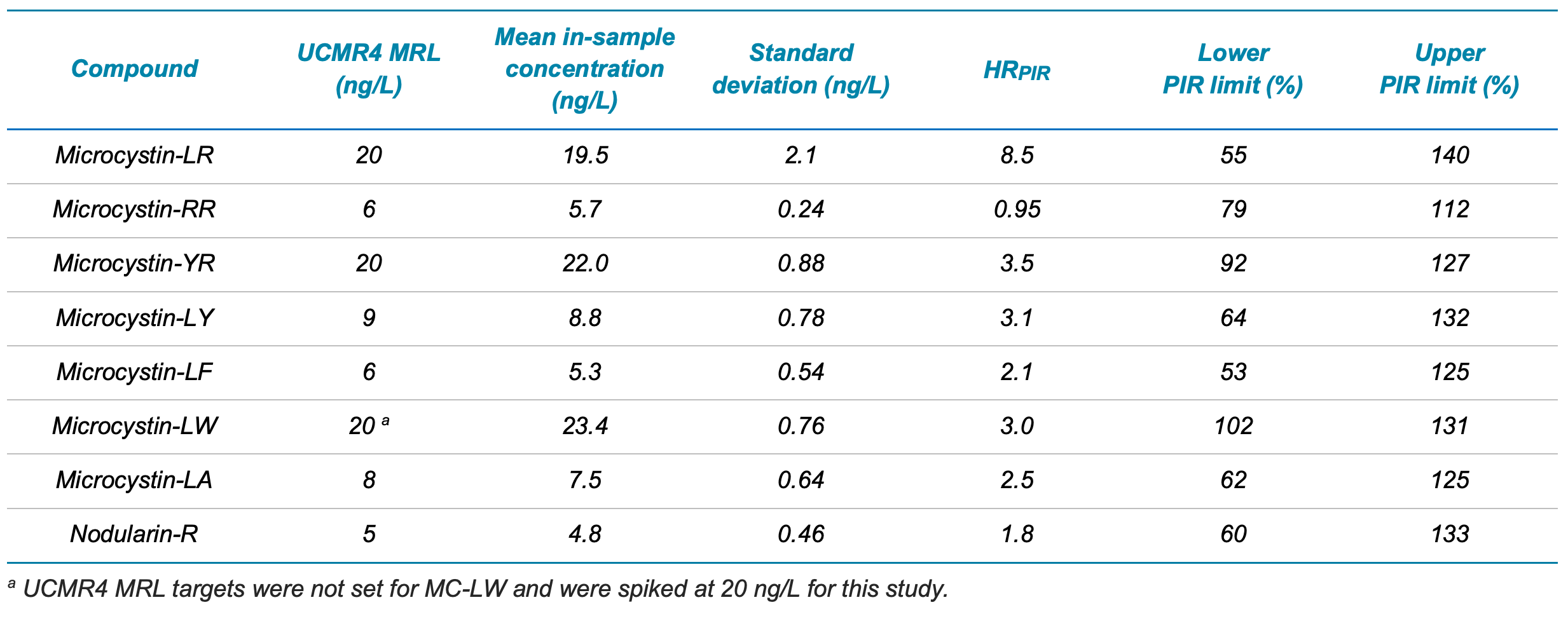
Figure 3. XICs of LRB and MRL LFB samples for MC-YR, MC-RR, MC-LR, MC-LY, MC-LF and MC-LW.
Conclusion
- EPA Method 544 IDC experiments necessary to demonstrate analytical proficiency, including low LRB background, LFB accuracy and precision and MRL verification were performed
- MRL confirmation was achieved at UCMR4 reporting levels ranging from 5 to 20 ng/L (0.005 to 0.02 µg/L)
- LFB spikes surpassed IDC criteria of ±30% and <30% for accuracy and precision, respectively
- Low ng/mL sensitivity of the QTRAP 4500 system was attained for microcystin analysis with solvent-based calibration standard LOQs ranging from 0.5 to 2.5 ng/mL
- Fast 10 min runtime showed good analyte separation and peak shape
References
- Method 544. Determination of microcystins and nodularin in drinking water by solid phase extraction and liquid chromatography/tandem mass spectrometry (LC/MS/MS). United States Environmental Protection Agency, Cincinnati, OH, February 2015. EPA Document No. EPA/600/R-14/474.
- Baliu-Rodriguez, D; Kucheriavaia,D; Palagama, DSW; Lad, A; O’Neil, GM; Birbeck, JA; Kennedy, JA; Haller, ST; Westrick, JA; Isailovic, D. (2020) Development and application of extraction methods for LC-MS quantification of microcystins in liver tissue. Toxins 12(4):263.