Protein quantification at sub-nanogram loads using Zeno SWATH data-independent analysis (DIA) and nanoflow chromatography
Using DIA-NN software and the ZenoTOF 7600 system
Ihor Batruch1, Alexandra Antonoplis2, Christie Hunter2
1SCIEX, Canada 2SCIEX, USA
Abstract
The combination of the ZenoTOF 7600 system with nanoflow chromatography enables reproducible protein and peptide identifications using very low amounts of sample. Here, a 300 nL/min flow rate was used with a 45-minute gradient to identify hundreds to thousands of proteins at loads of 5ng and below. Loads down to 250 pg on column were investigated to simulate protein amounts obtained when analyzing low numbers of cells. This workflow highlights the high sensitivity of the ZenoTOF 7600 system for Zeno SWATH DIA proteomics workflows.

Introduction
Data independent acquisition (DIA or SWATH DIA) has emerged as a comprehensive workflow for label-free quantitative proteomics, allowing for the acquisition of MS/MS spectra on all detectable peptides in an analysis. The ZenoTOF 7600 system is equipped to run Zeno SWATH DIA, which leverages Zeno trap activation on the platform for 5-6x increases in peptide MS/MS sensitivity in SWATH DIA workflows.1 Zeno SWATH DIA data can be interrogated using experiment-specific spectral libraries or libraries generated using in silico digestion approaches to identify and quantify proteins and peptides in the sample.2,3
The sensitivity gains provided by Zeno trap activation enable the use of nanogram-scale sample amounts on the platform for proteomics workflows. Recently, Zeno SWATH DIA was demonstrated to identify 3927 precursors at a 0.98 ng load of K562 cell digest and 7730 precursors at a 1.95 ng load of K562 cell digest with a 20 min microflow gradient (5 µL/min) and library-free data processing with DIA-NN.4
This work expands upon previous low-load experiments by leveraging nanoflow chromatography5 for enhanced protein and peptide detection. A 300 nL/min flow rate was used with 45 min gradients and a 15 cm nanoflow column for the analysis of sub-nanogram and nanogram amounts of K562 digest. Both library-based and library-free data processing strategies in DIA-NN were assessed for this workflow.
Figure 1. Proteins quantified with Zeno SWATH DIA and nanoflow chromatography. Transparent bars indicate identifications <1% global FDR and solid bars show identifications at <1% FDR and <20% CV. With a 300 nL/min flow rate and 45 min gradient for a 250 pg load, 880 proteins were identified at <1% global FDR and 327 proteins were identified at <1% FDR and <20% CV. Triplicate injections were analyzed for each load and data were processed with a library generated using the ZenoTOF 7600 system.2

Key features of Zeno SWATH DIA for protein identification at low sample loads
- Zeno trap activation provides ~5-6x increases in peptide MS/MS sensitivity and can be applied to MRMHR, SWATH DIA and data-dependent acquisition (DDA) workflows1-2
- The OptiFlow Turbo V ion source can be configured for nanoflow experiments in minutes, with probe and electrode positions fully optimized in the source6
- At sub-nanogram loading amounts, hundreds of proteins can be reproducibly quantified using Zeno SWATH DIA
- Library-free data processing can be used for Zeno SWATH DIA data analysis using DIA-NN software
Methods
Sample preparation: A digest of human K562 cell lysate from the SWATH performance kit (SCIEX) was used. The powdered digest was reconstituted in LCMS-grade water with 5% acetonitrile and 0.1% formic acid. This digest solution was then diluted with water and 0.1% formic acid to 50 ng/µL. For further dilutions to 0.25, 0.5, 1 and 5 ng/µL K562 digest, 5 fmol/µL bovine serum albumin (BSA) in 0.1% formic acid and water was used as a diluent. Sample loadings ranged from 250-5000 pg on column with a blank consisting of 5 fmol BSA on column. Sample vials were conditioned with 5 fmol/µL BSA digest prior to the addition of sample.
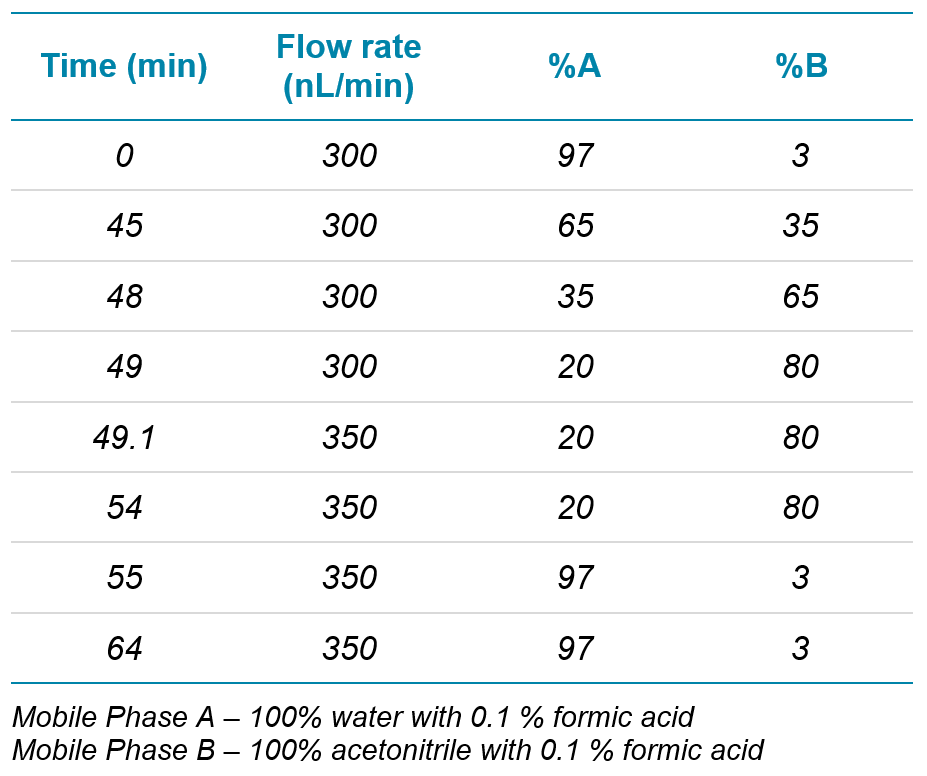
Chromatography: Separations were performed using a Waters ACQUITY UPLC M-class system plumbed for nanoflow chromatography (300 nL/min), using an Evosep analytical column (EV-1106 column,15 cm x 150 µm, 1.9 µm bead). All data were acquired using a 45 min gradient in direct injection mode with the gradient provided in Table 1.
Table 1. Gradient profiles used for nanoflow experiments.
Mass spectrometry: Data were acquired using a ZenoTOF 7600 system using variable window Zeno SWATH DIA with dynamic collision energy. Eighty-five variable windows (400-903 m/z) were implemented with an MS/MS accumulation time of 18 ms and an MS accumulation time of 50 ms. Source conditions for nanoflow experiments included an ion spray voltage of 3200 V, nano cell temperature of 225°C, Gas1 at 10 psi and CUR gas at 20 psi.
Data processing: Zeno SWATH DIA data were processed using DIA-NN software using both a spectral library generated on the ZenoTOF 7600 system and an in silico generated library. Data from 2 fractionation experiments of HeLa and K562 digests were each processed into 2 search results (HeLa and K562) in the ProteinPilot app in OneOmics suite for the library-based processing. The 2 search results for each cell line were then merged and retention time-aligned using the Extractor application in OneOmics suite to create a final ion library for Zeno SWATH DIA data processing.2 For the library-free processing, a spectral library was generated from a UniProt_Human FASTA (canonical+isoforms) file.7 This in silico library was used to process the DIA data to obtain protein identification numbers at 1% global FDR.8 Protein and peptide precursor areas from the *.pr_matrix.tsv and *.pg_matrix.tsv were pasted into Excel for computation of protein and peptide identifications at <20%CV.
Zeno SWATH DIA for protein identification at low loads
With the ZenoTOF 7600 system, MS/MS sensitivity increases ~5-6 fold for peptides with activation of the Zeno trap, which restores instrument duty cycle to >90%.1 This MS/MS acquisition mode can be applied to Zeno SWATH DIA workflows. Here, a range of low sample loadings were analyzed using Zeno SWATH DIA (Figures 1 and 2). A large ion library generated with fractionated HeLa and K562 cell digests and analyzed by Zeno DDA was used for data processing.2
At a 250 pg load of K562 digest, 880 proteins were identified at 1% global FDR, with 327 proteins quantified at 20% CV (Figure 1). Identifications increased as more sample was loaded for analysis, with 3752 proteins identified at 1% global FDR and 2457 proteins quantified at 20% CV at 5 ng on column. Triplicate injections were performed for each sample loading amount. As a control, a blank sample was prepared and analyzed using the same workflow as the other samples. No protein or peptides were detected in this sample, further validating the workflow.
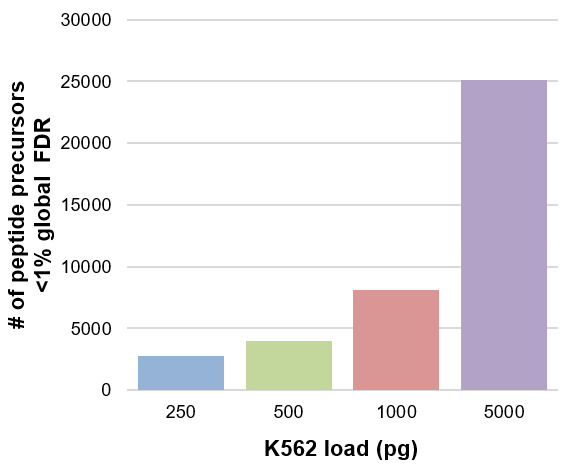
Figure 2. Peptide precursors at <1% global FDR with Zeno SWATH DIA and nanoflow chromatography. With a 300 nL/min flow rate and 45 min gradient for a 250 pg load, 2762 peptide precursors were identified at <1% global FDR. Triplicate injections were analyzed for each load and data were processed with a library generated using the ZenoTOF 7600 system.2
In addition to library-based processing, a library generated in silico from a human FASTA file was also used for data processing. Overall, a high degree of overlap was observed comparing the 2 processing approaches, with slightly more (~4%) protein identifications observed with library-based processing (Figure 3).
The inter-day reproducibility of low load experiments was evaluated by comparing 2 experiments performed 1 month apart. Different dilutions of the same K562 stock solution were prepared for each experiment and analyzed using Zeno SWATH DIA. Comparable identifications were observed at the 3 low loads (Figure 4).
Figure 3. Overlap of protein identifications at 1% global FDR between library-based and library-free processing at a 500 pg load. Most proteins identified at 1% global FDR were found using both processing approaches.
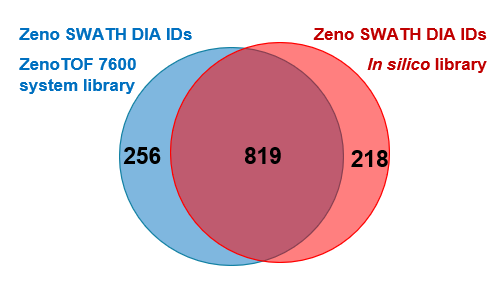
Figure 4. Comparison of 2 experiments performed with independent dilutions of K562 digest. Experiments were performed 1 month apart on the ZenoTOF 7600 system. Triplicate injections were performed for each loading amount in both experiments. Data were analyzed using library-based processing with the ZenoTOF 7600 system library in DIA-NN software. Transparent bars indicate identifications at <1% global FDR and solid bars show identifications at <1% FDR and <20% CV.

Conclusions
The combination of the ZenoTOF 7600 system with nanoflow chromatography enables reproducible protein and peptide identifications using very small amounts of sample. Here, a 300 nL/min flow rate was used with a 45-minute gradient. With triplicate injections, hundreds of proteins were identified at loads as low as 250 pg. This workflow highlights the sensitivity of the ZenoTOF 7600 system for Zeno SWATH DIA proteomics workflows.
References
- Large-scale, targeted, peptide quantification of 804 peptides with high reproducibility, using Zeno MS/MS. SCIEX technical note, RUO-MKT-02-13346-A
- Large-scale protein identification using microflow chromatography on the ZenoTOF 7600 system. SCIEX technical note, RUO-MKT-02-14415-A.
- Going library-free for protein identification using Zeno SWATH data-independent analysis (DIA) and in silico-generated spectral libraries. SCIEX technical note, RUO-MKT-02-14675-A.
- Ziyue W, et al. (2022) High-Throughput Proteomics of nanogram-scale samples with Zeno SWATH DIA. bioRxiv 2022.04.14.488299.
- Nanoflow Zeno SWATH data-independent analysis (DIA) for high-sensitivity protein identification and quantification. SCIEX technical note RUO-MKT-02-14798-A.
- Easy switching of sources and LC flow regimes on the ZenoTOF 7600 system. SCIEX community post, RUO-MKT-18-14697-A.
- Creating a library from a FASTA file for library-free data analysis. SCIEX community post, RUO-MKT-18-14611-A.
- Processing ZenoTOF 7600 system data with DIA-NN software. SCIEX community post, RUO-MKT-18-14611-A.