Abstract
In this technical note a new fragmentation type based on EAD1,2 is used to confirm the existence of disulfide and trisulfide linkages as well as free thiols in a bispecific mAb in which 2 different heavy chains and 2 identical light chains are linked. The data were acquired using a 10 Hz data-dependent acquisition (DDA) method and interpreted with Byos software (Protein Metrics Inc.). This workflow demonstrates a routine approach for the streamlined characterization of sulfide-linked peptides with a new level of data quality.
Introduction
The role of disulfide bonds is essential for maintaining tertiary and/or quaternary structures in proteins. Since the overall structure of proteins is related to their function, it is critical for the safety and efficacy of biotherapeutics to ensure that disulfide bond arrangements are as designed and expected. Existence of trisulfides can affect protein folding, structure and stability. In addition, they can further interfere with drug conjugation in case of cysteine-linked protein-drug conjugates. Characterization of linkage arrangements using bottom-up workflows has become increasingly popular due to advances in modern accurate mass spectrometry, novel fragmentation technologies and automated data processing software.3,4 However, sulfide bonds prevent effective fragmentation around linked cysteine residues when using collision-induced dissociation (CID), which limits the information available for identifying sulfide-bound peptides as well as sulfide patterns. Although alternative fragmentation mechanisms exist, these techniques can suffer from long reaction times, low sensitivity and lack of reproducibility.
The data presented in this technical note show a comprehensive analysis of linkage structure, including the confirmation of impurities, such as trisulfide linkages and free thiols. The analysis of linkage structures was performed using DDA of a non-reduced digested bispecific mAb with the ZenoTOF 7600 system (Figure 1). Novel signature fragments were identified to confirm the existence of trisulfides. With this workflow, standard and advanced characterization, leveraging a highly reproducible fragmentation technique based on EAD1,2, is achievable in 1 injection. This workflow enables a streamlined characterization accessible to every user level.
Key features of the ZenoTOF 7600 system
- Confident confirmation of trisulfide-linked peptides: The EAD platform method5 provides signature fragments to differentiate disulfide and trisulfide linkages, enabling a higher level of structural characterization in 1 injection
- New depths of peptide mapping analysis: EAD with fast DDA enables alternative fragmentation for routine, in-depth analysis of next-generation protein therapeutics and standard mAbs
- Higher MS/MS sensitivity: Increased detection of fragments (by 5- to 10-fold) using the Zeno trap enables higher confidence in data assignment, which enables free thiol detection at low abundances
- High reproducibility: Reproducible fragmentation with EAD for singly, doubly and multiply charged ions enables analysis of more precursors than other alternative and low-reproducibility fragmentation techniques
- Streamlined and easy to use: Fully automated data acquisition in DDA mode using EAD with SCIEX OS software and automated data interpretation with Byos software (Protein Metrics Inc.) simplifies the entire user experience

Methods
Sample preparation: The bispecific mAb sample was prepared using the AccuMap Low pH Protein digestion kit (Promega) under non-reducing condition.
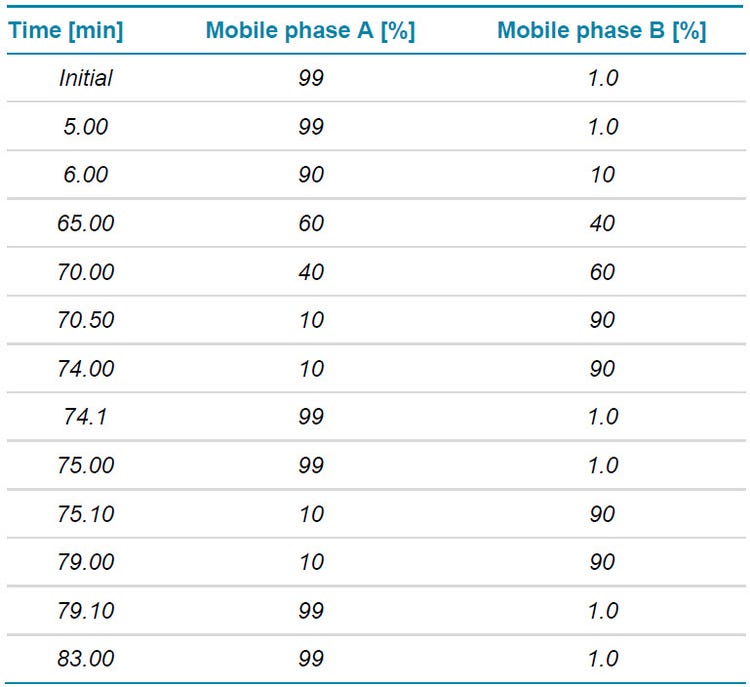
Chromatography: A total of 3 µL (4 µg) of the Lys-C digest were separated with a Waters ACQUITY CSH C18 column (2.1 × 100 mm, 1.7 μm, 130 Å) using an ExionLC AD system. The mobile phase (A) consisted of water with 0.1% formic acid, while the organic phase (B) was acetonitrile with 0.1% formic acid. A gradient profile was used at a flow rate of 250 μL/min (Table 1). The column temperature was maintained at 50°C.
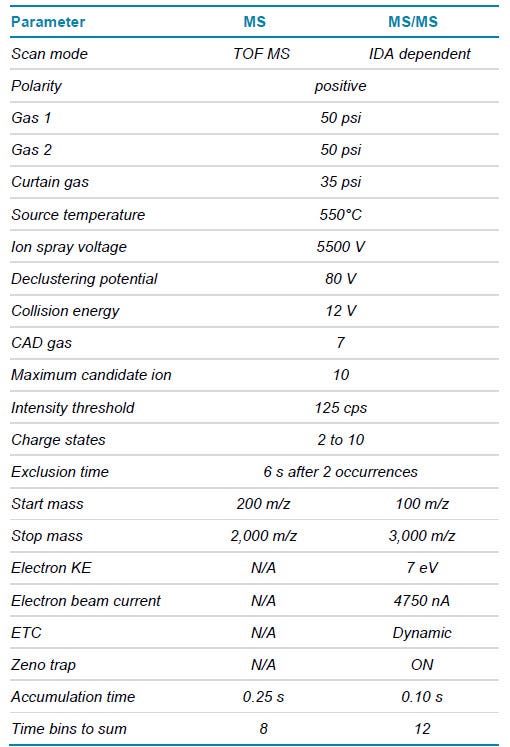
Overview
Traditional antibody-based therapeutics consist of 2 light chains and 2 heavy chains with multiple interchain and intrachain disulfide bonds to maintain a protein’s 3-dimensional structure. Misfolding can lead to a change in protein function such as antigen recognition or binding affinity6 and can affect both the efficacy and safety of a biotherapeutic. With the advancement of modern mass spectrometry, bottom-up approaches have become the method of choice for the characterization of multiple product quality attributes, enabling the simultaneous identification and localization of modifications, including characterization of disulfide bonds.4,7 The most used fragmentation approach for this workflow (CID) typically leads to peptide backbone fragments while leaving the disulfide bonds intact. This can result in a complicated, yet incomplete MS/MS spectrum that is difficult to interpret. In addition, CID struggles to obtain high fragment coverage for long disulfide-linked peptides, especially around the residues close to the cysteine linkages. Confidently identifying low-abundance disulfide scrambling events can therefore be challenging with CID.
While alternative fragmentation techniques have been used for these analyses in the past, their adoption by the biopharmaceutical industry has been limited by their overall low sensitivity and lack of automated DDA workflows.8-10
Disulfide-bondes peptides
The study presented here focused on the detailed characterization of complex sulfide linkages in a multispecific mAb using the ZenoTOF 7600 system. To minimize disulfide scrambling introduced during sample preparation, all free cysteines were capped with N-ethylmaleimide (NEM) followed by digestion with Lys-C using a low pH digest kit. The low pH minimizes any rearrangements of disulfide bonds during the digestion procedure. Fragmentation and identification data from DDA with either Zeno EAD or Zeno CID were compared. Information gained from this comparison was used to establish a routine method for complicated linkage determination. Both EAD and CID were able to identify all expected disulfide linkages from the bispecific mAb, while EAD provided a higher MS/MS coverage for long peptides.
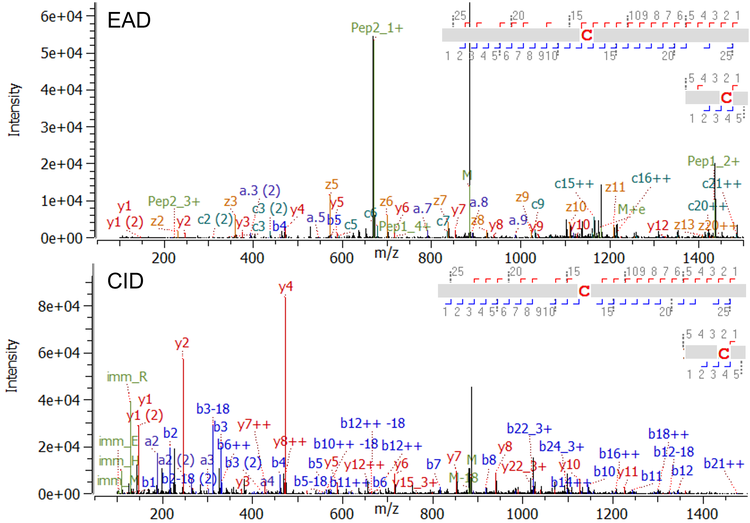
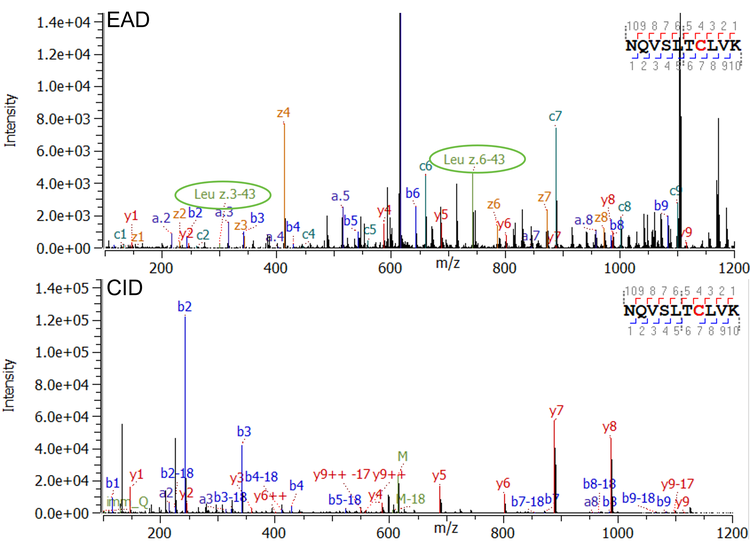
Figure 2 shows an example of typical EAD and CID MS/MS spectra for a regularly sized disulfide-linked peptide with a molecular weight (MW) of 3,536 Da. Both, EAD and CID, achieved a high MS/MS coverage for this linked peptide (97% for EAD and 90% for CID). Among the dominant fragments observed in the EAD spectrum are the charge states +1 and +2 of the 2 corresponding peptides (green labels of peptide 1 and peptide 2 in Figure 2, top) derived from cleavage of the disulfide bond with EAD. That additional level of information obtained by EAD enables confirmation of the identity of the connected peptides, ruling out potential misassignments, especially in the case of low-abundance scrambled disulfide bonds. Figure 3 demonstrates an example of EAD and CID MS/MS spectra from a large and highly charged disulfide-linked peptide at m/z 952.617 with z = +8 and a MW of 7,612 Da. While CID is effective in fragmenting the termini of the connected peptides, the technique struggles to effectively dissociate the peptide backbones towards the center of the long sequences (red circles in Figure 3 right, bottom).
Identification of free thiols
Correct disulfide assembly is essential for protein function. However, free thiols are frequently observed in protein therapeutics7 in low abundance. These patterns can result from incomplete processing during expression, or chemical cleavage under reducing conditions.8 To accurately identify existing free thiols, NEM is used as a capping agent to protect all existing free cysteines before enzymatic digestion. The capping agent introduces a 125 Da increase of the MW. Here, a low amount of free thiols were detected at different cysteine sites of the bispecific mAb sample. Figure 4 is an example of a peptide containing a free cysteine in the conserved region of the heavy chain of the bispecific mAb. Both Zeno EAD (Figure 4, top) and Zeno CID (Figure 4, bottom) provided highly descriptive spectra with full fragment coverage, which confirm the existence of a free thiol at the cysteine in this peptide. The fragments c7, z4 in the EAD spectrum and y4, y5, etc. in the CID spectrum all show a +125 amu increase, which can be attributed to the NEM capping of the free thiol. Besides the high level of sequence confirmation, EAD spectra also offer the ability to differentiate leucine (Leu) from iso-leucine (Ile) as shown in detail previously.9 In this particular peptide, 2 Leu residues present based on the theoretical sequence. Fragments z6-43 and z3-43 were detected in the MS/MS spectrum (encircled fragments in Figure 4, top), which verify that these 2 amino acids are both Leu. No evidence for Ile was found for this peptide.
Trisulfide-bonded peptides
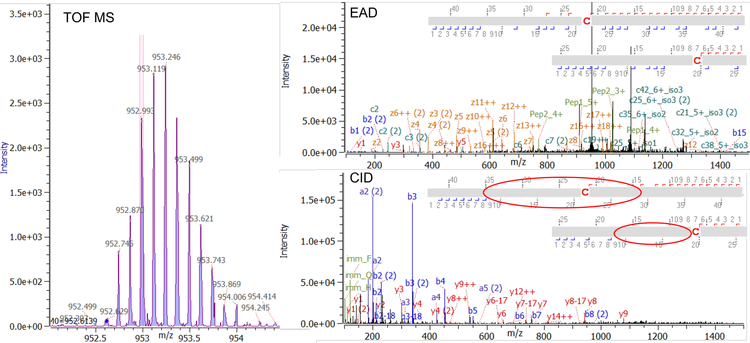
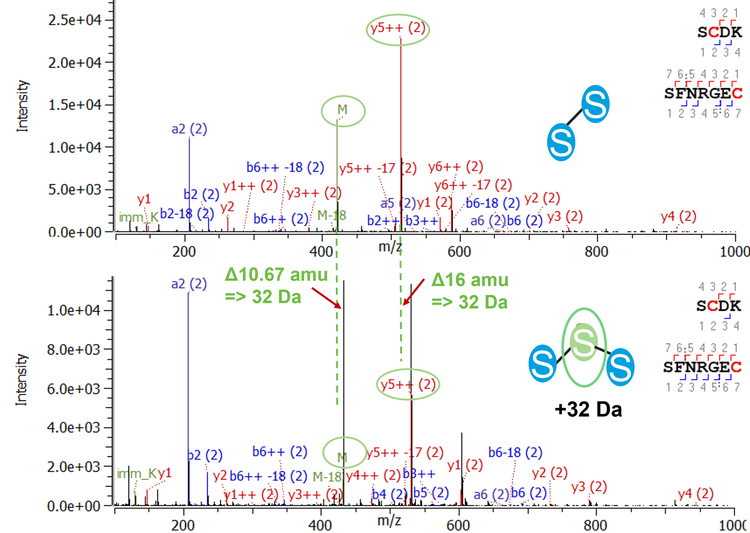
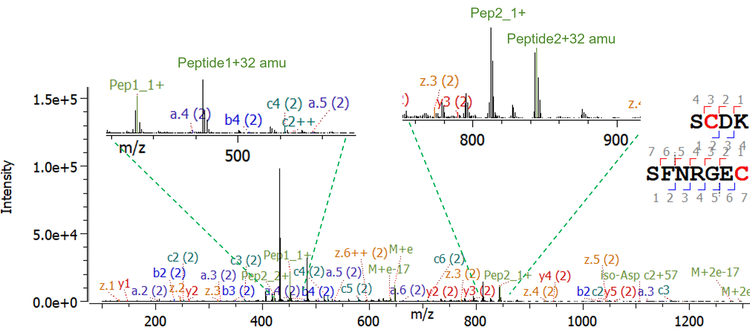
Trisulfide bonds are formed by the insertion of an additional sulfur into a disulfide bond, causing a shift of the MW 32 Da. Although it is reported that trisulfides have no effect on antigen binding or on the stability of the therapeutic molecule6, they are known to change protein folding and may cause challenges in case of additional bioengineering processes. Therefore, the identification and close monitoring of this post-translational modification is necessary. A high abundance of trisulfides was detected in the studied molecule at the interchain disulfide bond between heavy and light chains. As expected for trisulfide-bonded peptides, a 32 Da increase was observed for the MW of the precursor compared to the disulfide-bonded peptide. Further evidence was found using MS/MS information. Figure 5 shows the comparison of the MS/MS spectra of the disulfide- and trisulfide-containing peptide using CID. As the precursor is triply charged, a 10.67 amu increase was observed in the spectrum of the trisulfide-bonded peptide. Meanwhile the y5++ fragments presented a 16 amu shift. The EAD spectrum for the trisulfide containing peptide is shown in Figure 6. As previously discussed, EAD can dissociate disulfide bonds, resulting in signals for the 2 cleaved peptides in the MS/MS spectrum.10 Furthermore, EAD can dissociate trisulfide bonds, leading to 4 different fragments: peptide 1, peptide 1+sulfur, peptide 2, peptide 2+sulfur (Figure 7). While the signals for peptide 1 and peptide 2 in the MS/MS spectrum give further confidence in the identity of both linked peptides, the fragments of the respective peptide with additional sulfur can be used as diagnostic ions to confirm the existence of trisulfide bonds with very high confidence. Neither of these fragments can be obtained by CID.

Conclusion
- The existence of trisulfide bonds was confidently confirmed through signature ions generated by DDA with Zeno EAD: a 1-injection platform method without the need for optimization
- Confident sequence and disulfide linkage confirmation of long disulfide-bonded peptides were achieved with EAD, showing superior fragmentation coverage compared to CID
- Free thiols were detected at low levels with high confidence using EAD, even in case of low-charged precursors
- In addition to studying sulfide arrangements, amino acid isomers can be differentiated with the same Zeno EAD data, enabling high efficiency to obtain a wealth of information on the analyte of interest
- Automatic, state-of-the-art data processing enables the routine and advanced characterization of complex biotherapeutics and standard mAbs in a reproducible manner using Byos software from Protein Metrics Inc.
References
- Baba, T. et al. (2015) Electron capture dissociation in a branched radio-frequency ion trap, Anal. Chem. 87(1), 785−792.
- Baba, T. et al. (2021) Dissociation of biomolecules by an intense low-energy electron beam in a high sensitivity time-of-flight mass spectrometer. J. Am. Soc. Mass Spectrom.32(8), 1964–1975.
- Srebalus Barnes, C.A. et al. (2007) Applications of mass spectrometry for the structural characterization of recombinant protein pharmaceuticals. Mass Spectrom. Rev. 26(3), 370–388.
- Lakbub, J.C. et al. (2018) Recent mass spectrometry-based techniques and considerations for disulfide bond characterization in proteins. Anal. Bioanal Chem. 410(10), 2467–2484.
- An evaluation of a single injection platform method for advanced characterization of protein therapeutics using electron activation dissociation (EAD). SCIEX technical note, RUO-MKT-02-13965-A.
- Gu, Sheng, Pepinsky R et al. (2010) Characterization of trisulfide modification in antibody. Anal Biochem 400(1):89-98.
- Devarie Baez N, Furdui C et al. (2015) Mass Spectrometry in Studies of Protein Thiol Chemistry and Signaling: Opportunities and Caveats. Free Radic Biol Med. 80: 191-211.
- Chapter 1.4 Analysis of Disulfide Bond Formation in Therapeutic Proteins. Oxidative folding of proteins: basic principles, Cellular Regulation and Engineering, 2018.
- Comprehensive peptide mapping of biopharmaceuticals utilizing electron activation dissociation (EAD). SCIEX technical note, RUO-MKT-02-12639-B.
- Confirmation of disulfide linkages in adalimumab using electron activated dissociation (EAD). SCIEX technical note, RUO-MKT-02-12913-B.
Frequently asked questions
Why is confident characterization of linkage structures critical for bispecific monoclonal antibodies?
Bispecific monoclonal antibodies contain more complex architectures than conventional mAbs, often involving multiple heavy chain arrangements and engineered cysteine linkages. Because disulfide bond patterns play a central role in maintaining correct higher order structure, unanticipated linkage variants can impact stability, function, and developability. This technical note highlights the need for analytical workflows that can confidently confirm expected disulfide structures and detect impurities such as trisulfides or free thiols in complex bispecific mAb formats.
What linkage related attributes can be characterized using this EAD‑based LC‑MS/MS workflow?
The EAD‑based workflow demonstrated in this technical note enables confirmation of disulfide and trisulfide linkages as well as identification of free thiols in a bispecific mAb composed of two different heavy chains and two identical light chains. By applying bottom‑up LC‑MS/MS analysis under non‑reducing conditions, the workflow provides detailed insight into sulfide‑linked peptides and impurity patterns that are difficult to resolve using traditional CID‑based methods.
How does this workflow support analytical development scientists working on complex mAb formats?
Analytical development scientists must confirm that engineered antibody architectures match their intended design and remain consistent across development stages. This technical note demonstrates a single‑injection, data‑dependent acquisition (DDA) workflow that enables routine characterization of complex linkage structures using EAD. By simplifying sulfide‑linkage analysis and producing high‑quality, interpretable data, the approach helps development teams reduce ambiguity, detect impurities earlier, and generate more confident structure‑related conclusions for bispecific mAb candidates.
How does EAD overcome the limitations of CID for sulfide‑linked peptides?
CID does not cleave the disulfide bond and often fails to produce informative fragmentation near cysteine residues involved in disulfide or trisulfide bonds, limiting confidence in linkage assignment. In this workflow, EAD provides an alternative fragmentation mechanism that cleaves both disulfide bond and peptide backbone, generating signature fragments around sulfide linkages. This enables direct confirmation of linkage structures that are otherwise difficult to elucidate, improving confidence in peptide‑level structural interpretation for complex antibodies.
How are trisulfide linkages and free thiols detected using this EAD‑based approach?
The technical note shows that EAD fragmentation produces characteristic fragment ions that distinguish trisulfide‑bonded peptides from conventional disulfide linkages and enables identification of free thiols as structural impurities. By applying EAD to non‑reduced peptide digests in a DDA workflow, these linkage variants can be detected and confirmed within a single analysis, providing deeper insight into antibody linkage heterogeneity.
Why is achieving comprehensive linkage characterization in a single injection valuable?
Traditional linkage‑analysis workflows often require multiple injections, complementary methods, or specialized expertise to piece together sulfide‑bond information. In this technical note, comprehensive characterization of disulfide bonds, trisulfides, and free thiols was achieved in a single LC‑MS/MS injection using EAD. For analytical development scientists, this streamlined approach reduces method complexity, improves reproducibility, and makes advanced linkage characterization more accessible across different user experience levels.