Characterization of the multi-acetylated sites of human histone 2A variants by CESI with SWATH acquisition
Using the CESI 8000 Plus ESI-MS system coupled to the TripleTOF 6600+ system
Zuzana Demianova1, Zhichang Yang1, Klaus Faserl2, Bettina Sarg2, Sahana Mollah1 and Herbert Lindner2
1SCIEX, USA; 2Division of Clinical Biochemistry, Innsbruck Medical University, Innsbruck, Austria
Abstract
Histones have high sequence homology and are heavily modified by dynamic post translational modifications (PTMs) making characterization challenging. Capillary electrophoresis (CE) separates analytes by size to charge ratio with high separation efficiency and sensitivity and can be used as an alternative strategy to LC when a higher separation resolving capability is needed. Here, characterization of histone peptides containing multiple acetylation sites using CESI and SWATH acquisition was performed. Highlighted are two peptides of histone 2A (H2A) variants with two and three acetylation sites where selected. CESI with SWATH acquisition was able to separate and identify positional isomers with MS/MS confirmation.

Introduction
Histones are heavily modified by dynamic post translational modifications (PTMs) which can affect the structure of chromatins and regulate DNA transcription and replication.1,2 Separation of isomeric histone tryptic peptides can be challenging using liquid chromatography (LC) due to similar hydrophobicity, especially when the PTM sites are nearby.3 Capillary electrophoresis (CE) separates analytes by size to charge ratio and is known for its high separation efficiency and sensitivity. Hence CE can be used as an alternative strategy to LC when a higher separation resolving capability is needed.
Histone H2A is one of the five histone proteins that is involved in function-dependent dynamic organization of chromatin. Among core histones, the H2A family exhibits highest sequence divergence, resulting in the largest number of variants known. They often vary by only a few amino acids differing from each other, mostly at their C-terminus. Depending on H2A type and biological context, H2A proteins also undergo a variety of modification reactions like phosphorylation, acetylation, ubiquitination etc., which have striking biological implications. But the wide range of possible modifications coupled with the high sequence similarity makes the detailed understanding of this protein family analytically challenging.
Here, histone H2A peptides containing multiple acetylation sites have been characterized using CESI for separation and a SWATH acquisition strategy for identification/quantification. Highlighted are two peptides of histone 2A (H2A) variants with two and three acetylation sites. CESI was able to separate the positional isomers (Figure 1) and MS/MS was used for site confirmation. CESI-MS was demonstrated as a powerful tool for comprehensive identification of the histone family.
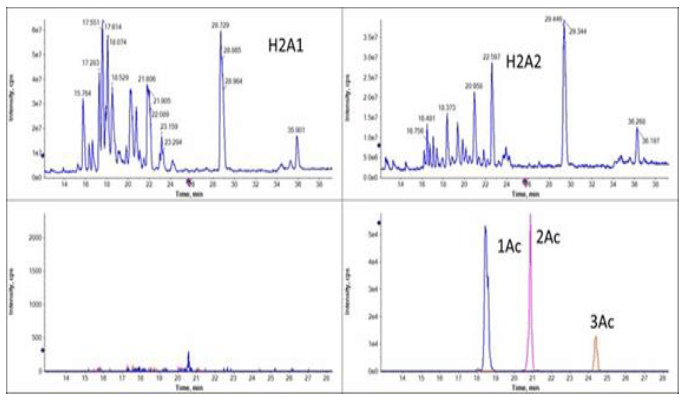
Figure 1. Separation of multi-acetylated peptides unique for H2A2 fraction. The top panels show total ion electropherogram (TIE) of fraction H2A1 and H2A2. The bottom panels show extracted electropherograms of the multi-acetyl modified peptide AGGKAGKDSGKAKTKAVSR.
Key Features of CESI-MS for histones
- Separation of peptides with different/multiple post-translational odifications (PTM) by their electrophoretic mobility
- Separation and identification of site-specific acetylation on the same peptide of histone 2A variants
- High sensitivity of CESI allows for identification of low-level PTMs
- Minimum sample required (nanoliters) to obtain high sequence coverage of very challenging histone variants
- SWATH acquisition on the TripleTOF 6600+ system for acquisition of comprehensive, high quality MS/MS spectra for peptide identification and PTM site localization.
Methods
Sample preparation: Nuclei from human lymphoblastic T-cells (line CCRF-CEM) were extracted with 0.2 M sulfuric acid. Histones present in the supernatant were precipitated by TCA at a final concentration of 20% for 1 hour on ice. The pellet obtained by centrifugation was washed with acidified acetone. Proteins were then re-dissolved in 0.1% (v/v) β-mercaptoethanol and separated by RP-HPLC using a C4 column (250 x 3mm; 5μm particle pore size; 30 nm pore size) and an acetonitrile gradient with the addition of 0.1% TFA (v/v). Obtained fractions were lyophilized.
Table 1. CESI conditions were used to separate the histone 2A variants.
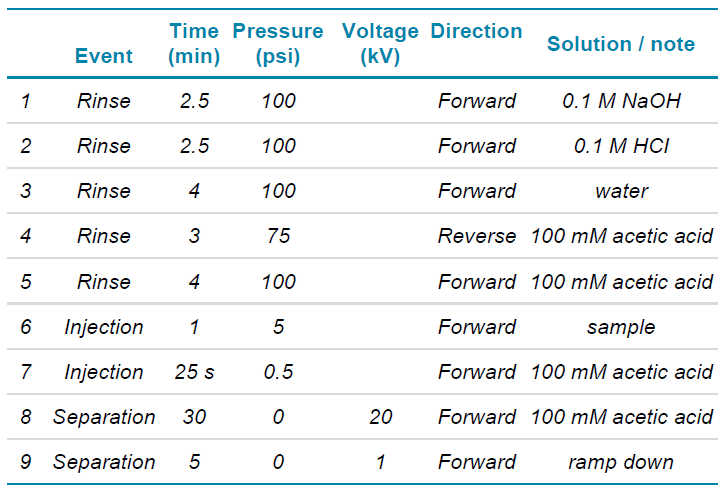
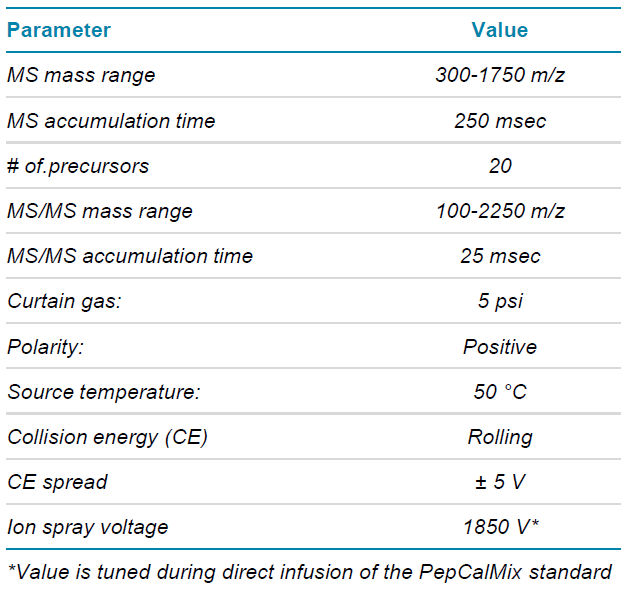
Table 2. DDA mass spectrometry parameters.
For enzymatic digestion, human core histones were dissolved in ammonium bicarbonate buffer (100 mM, pH 8.0) and digested overnight at 37°C using endoproteinase Glu-C at a ratio of 1:20. Resulting peptides were lyophilized and re-dissolved in 50 mM ammonium acetate buffer (pH 4.0) to a concentration of 1μg/μL. For more details see reference 4 and 5. Prior analysis, lyophilized samples (5 μg) were reconstructed to 5 μL with 50 mM ammonium acetate, pH 4.
Instrument configuration: The CESI 8000 Plus system with the silica surface OptiMS cartridge (SCIEX, P/N B07367) was coupled with the TripleTOF 6600+ system through a NanoSpray III source and a CESI adapter (SCIEX, P/N B07363). 32 Karat software and Analyst TF software 1.8.1 were used to operate the CESI system with the TripleTOF 6600+ system.
Capillary electrophoresis and mass spectrometry: The separation conditions are listed in Table 1. The sample was injected hydrodynamically at 5 psi for 60 sec, resulting in approximately 44 nL injected volume, which is about 220 ng of total protein content on the cartridge. The BGE (100 mM acetic acid) was prepared freshly on the analysis day.
MS method parameters: The histone 2A variants samples were analyzed by data dependent acquisition (DDA) and data independent acquisition (SWATH acquisition) methods. The TOF MS mass range for both acquisitions were set 300-1750 m/z, with a cycle time of approx. 0.7 sec.
Table 3. SWATH acquisition mass spectrometry parameters.
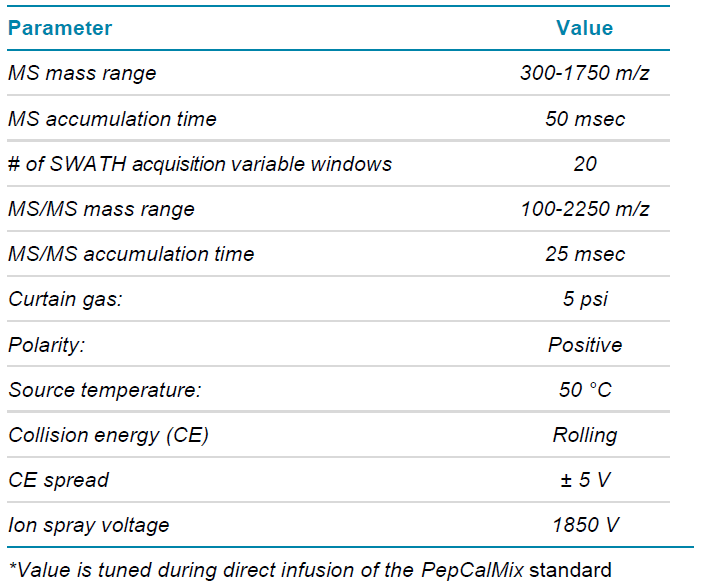
Table 4. Identified proteins with high sequence coverage in the fraction H2A1.
Method details are listed in Table 2 for DDA acquisition and Table 3 for SWATH acquisition. The SWATH acquisition method was built using 20 variable Q1 windows, and the window widths were determined based on the MS ion intensity distribution using the SWATH acquisition variable window calculator.6
Data processing: SCIEX OS software was used to visualize and analyze the data. ProteinPilot software was used for protein identification from DDA datasets using UNIPROT KB database from March 2021 (uniport.org) with special factors, purified histones, and human species.
Biological importance of histone variants
The H2A family of histones exhibits the highest sequence divergence. In UniProt database, 18 different entries for human H2A histones can be found. H2A variants are ideal candidates to control access to the DNA as they are located at the entry and exit positions along the wrap of nucleosomal DNA. This might also explain the involvement of distinct H2A variants in most diverse biological processes. H2A.X, for example, has been shown to be involved in the DNA damage response.7 H2A.Z has the lowest sequence homology (60%) compared to canonical H2A proteins in a given species, but between different species, H2A.Z is highly conserved with a given sequence similarity of ~80%.8 These data suggest that H2A.Z fulfils specific and unique functions that cannot be carried out by other H2A variants. It is also important to note that in cancer, studies have revealed the up- and down-regulation of a variety of H2A isoforms like H2A type 1, H2A1 A, H2A1 B/E, H2A1 C, H2A1 D, etc.9 Therefore, determining both the peptide sequence and the location of key PTMs is crucial in the study of histone biology.
Protein identification
In this experiment, SWATH acquisition was used to generate the MS/MS data. With SWATH acquisition, all ionized tryptic peptides of a given sample that fall within a specified m/z window are fragmented to generate MS/MS spectra in an unbiased fashion,10 thus providing a more complete protein coverage. As seen in Table 4, although the major component in fraction H2A1 is H2A type 1-D, there are also presence of other H2A variants and to a smaller extend H4 and H3.2 While present at lower levels, a good sequence coverage of other variants was obtained with identification of multiple peptides per protein. Similarly, with H2A2 fraction, other histone variants were detected with high sequence coverage, along with the major component H2A type 2-A as shown in Table 5.
Separation of acetylated species with CESIMS
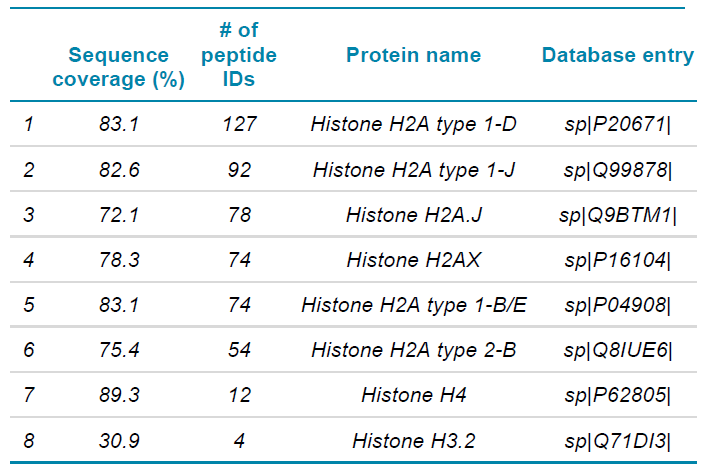
CESI-MS can differentiate isomeric histone tryptic peptides, which can be a challenge when using LC-MS. Peptide GKQGGKAR is mono-acetylated at either K2 or K6 site. With CE separation, the two mono-acetylated peptides were well separated as shown in the top panels of Figure 2. Quantification information of the isomeric peptides can be achieved from electropherograms of the precursor ion. The site-specific fragments (228.134 m/z and 530.311 m/z) from the MS/MS spectrum further confirmed the separation and identification of the two isomeric peptides.
Figure 2. Characterization of acetylated peptide GKQGGKAR. Top panels show an electropherogram of the separation of mono-acetylated peptide using CESI-MS and position-specific fragments (228.134 m/z and 530.311 m/z). Bottom panels show no separation of the two mono-acetylated peptides using LC separation. Both signature fragments (228.13 m/z and 530.30 m/z) were detected in the MS/MS spectrum.
On the other hand, it was challenging to separate the isomeric peptides with LC where the peaks are co-eluting at 7.78 min (Figure 2, bottom). The confirmation of the presence of both species was able to be made only because site-specific fragments (228.13 m/z and 530.30 m/z) for both species were found in the MS/MS spectrum.
Using this combination of CESI with SWATH acquisition also enabled the localization of acetylation present on different sites on the peptide AGGKAGKDSGKAKTKAVSR. This sequence is found on both the H2A1 and H2A2 variants. The peptide was found to be multi-acetylated in H2A2 fraction, but no acetylation form was identified in H2A1 fraction (Figure 1). Peptides in H2A2 with different acetylation level (1Ac-, 2Ac- and 3Ac-) were clearly separated (Figure 3, top) by the CESI separation with identification confirmed from the MS/MS analysis (Figure 3, bottom).
Peptide GKQGGKAR with different levels of acetylation were also well separated and identified by CESI-MS as shown in Figure 4. Acetylation on lysine reduces peptide charge and leads to reduction of electrophoretic mobility of peptides during CE separation.11 The electropherograms shown in Figure 3 and Figure 4 observe this rule in which higher acetylation level leads to slower migration. The relative abundance of the peptides with different acetylation level can also be determined from the MS1 precursor ion spectra. Mass spectra corresponding to the peptides with 4 different acetylation status were shown in the bottom panels of Figure 4.
Figure 3. Characterization of the acetylation of peptide AGGKAGKDSGKAKTKAVSR in H2A2. The top panel shows separation of peptide with different acetylation (1Ac-monoacetyl, 2Ac-diacetyl, 3Ac-triacetyl. The bottom panels show the MS/MS spectrum of peptides containing K4(1Ac), K4 and K11(2Ac); K4, K7 and K11 (3Ac) to confirm the identity of the peptides.
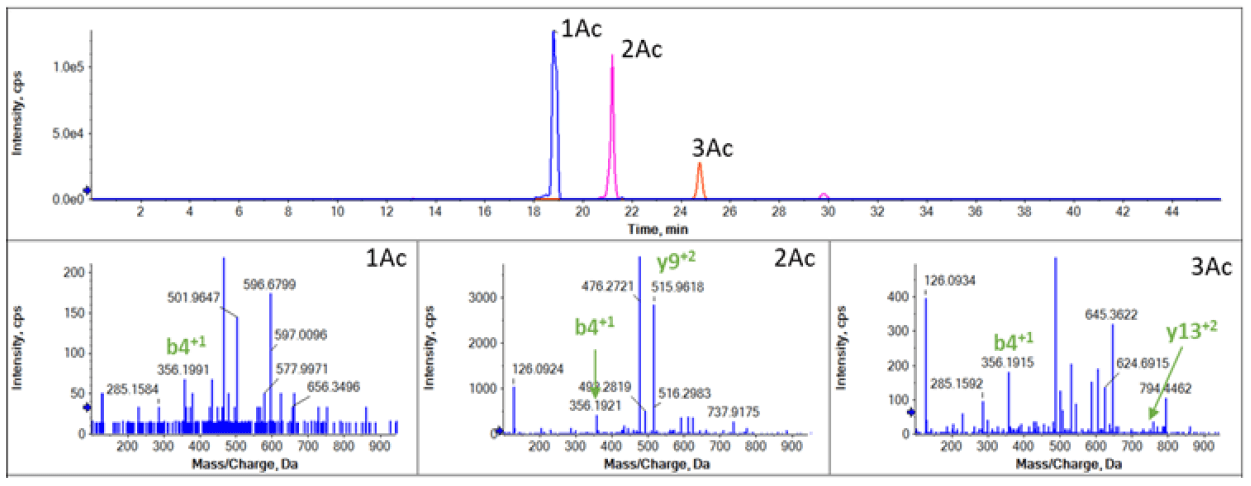
Figure 4. Acetylation sites of the GKQGGKAR peptide in H2A1 and H2A fractions. The top panel shows XICs of MS fragments. The bottom panels show precursors mass of peptide with/without modification.

Conclusions
- CESI-MS enabled the separation of isomeric acetylated peptides of histone, which can be difficult to separation by LC-MS system. MS/MS spectra was used to confirm the identity of the peptides and the acetylation sites.
- Using SWATH acquisition as the MS acquisition technique ensured identification of very challenging H2A variants with high sequence coverage
- Inherent sensitivity of CESI-MS enabled analysis from only nanoliters of sample
- Peptides with different level of acetylation (1Ac-, 2Ac- and 3Ac-) were well separated and identified by CESI-MS. The relative abundance of the acetylation variants was determined from the MS1. Even low-level acetylation can be identified using this highly sensitive platform.
References
- Xu D, et al. (2009) Covalent modifications of histones during mitosis and meiosis. Cell Cycle. 8(22): 3688- 94.
- Jung HR, et al. (2013) Precision mapping of coexisting modifications in histone H3 tails from embryonic stem cells by ETD-MS/MS. Anal Chem. 85(17): 8232-9.
- Singer D, et al. (2010) Separation of multi-phosphorylated peptide isomers by hydrophilic interaction chromatography on an aminopropyl phase. Anal Chem. 82(15): 6409-14.
- Lindner H, et al. (1986) Histone Separation by High- Performance Liquid Chromatography on C4 Reverse-Phase Columns. Anal. Biochem., 158, 424-430.
- Sarg B, et al. (2013) Comparing and combining capillary electrophoresis electrospray ionization mass spectrometry and nano-liquid chromatography electrospray ionization mass spectrometry for the characterization of post-translationally modified histones. Mol Cell Proteomics, 12(9): 2640-56.
- Download the SWATH acquisition Variable Window Calculator - Excel tool. Download from.
- Podhorecka M, et al. (2010) H2AX Phosphorylation: Its Role in DNA Damage Response and Cancer Therapy. J Nucleic Acids. 920161.
- Nissen KE, et al. (2017) The histone variant H2A.Z promotes splicing of weak introns. Genes Dev. 31(7): 688- 701.
- Shah S, et al. (202) Histone H2A isoforms: Potential implications in epigenome plasticity and diseases in eukaryotes. J Biosci., 45: 4.
- Ludwig C, et al. (2018) Data-independent acquisition-based SWATH-MS for quantitative proteomics: a tutorial. Mol Syst Biol. 14(8): e8126.
- Chen D, et al. (2020) Predicting Electrophoretic Mobility of Proteoforms for Large-Scale Top-Down Proteomics. Anal Chem. 92(5): 3503-3507.