ProteinPilot™ Report for ProteinPilot™ Software
Detailed analysis of protein identification / quantitation results automatically
Sean L Seymour, Christie Hunter
SCIEX, USA
Abstract
In addition to providing lists of identified proteins and peptides, database search results from ProteinPilot™ Software contain a wealth of additional information that can be analyzed post-acquisition. Whether mining to assess results quality, obtaining additional quantitative information or performing LC-MS workflow improvements, easy access to this extra information is of high value. This analysis is automated in the ProteinPilotReport.xlsx, which is generated after every ProteinPilot software search. In the large report, there are over 20 detailed dashboards that help characterize the LC-MS results, including sample preparation, chromatographic quality, MS acquisition quality, and many others.

Introduction
Powerful mass spectrometers like the TripleTOF® 6600 and 5600 Systems can rapidly generate extremely large amounts of data. For today’s researchers, tools that can logically and efficiently distill the massive amounts of data down into easily interpretable results are critical. ProteinPilot Software is a powerful, robust, easy to use software tool for protein identification and quantification for discovery research and protein characterization1. With its hybrid sequence tag and database search approach using feature probabilities, the powerful Paragon™ Algorithm can search for hundreds of modifications and sequence variants in a single search2. Coupled with the Pro Group™ Algorithm for protein inference analysis, peptide results are condensed down to the most defensible set of detected proteins with ambiguity among multiple accession numbers reported when appropriate.
In addition to identification and quantitation information, there are many different types of post-acquisition analysis that can be performed that are highly valuable to the protein researcher to ensure results quality and enable workflow refinement. Many of these types of analysis have been combined into a single Excel-based processing tool, the ProteinPilot Report.
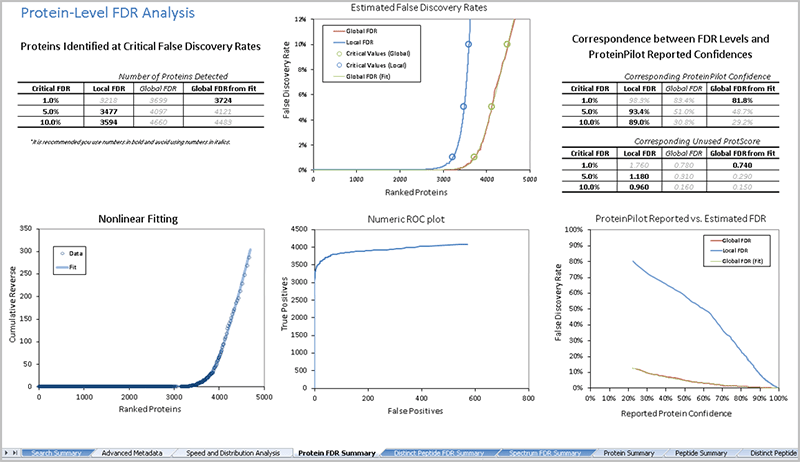
Figure 1. False discovery rate (FDR) analysis. FDR analysis should be performed when large scale protein identification is being performed. A detailed report is generated for every database search.
Key features of the ProteinPilot Report
- Automatic generation of a Report with every search
- Small report contains detailed meta data, FDR analysis, data exports
- In addition, the large report contains over 20 dashboards of valuable information5
- Enables the rapid assessment of the quality of identification and quantification.
- Enables the characterization of sample preparation – digestion quality, modification frequencies, labeling efficiency, etc.
- Enables the optimization of acquisition parameters using detailed metrics on acquisition redundancy, chromatography, mass accuracy, etc.
- Generate volcano plots and compute false discovery rates of differential expression for simple quantitation studies.
- Virtually all quantitative metrics (>7000 data points) are captured in a single column that can be saved for future use –from simple comparison to complex data mining.
Basic reporting for ease of publication
For every ProteinPilot Software database search, a detailed false discovery rate (FDR) analysis is performed and a rigorous report is generated, detailing the quality of protein and peptide identifications3. FDR analysis is performed at the spectral, peptide and protein level (Figure 1). A novel non-linear fitting method is used to determine both a global and a local FDR from the decoy database search3.
A detailed meta-data report is generated which contains a large amount of information that is useful for reporting search details for publication.
Characterization of MS acquisition
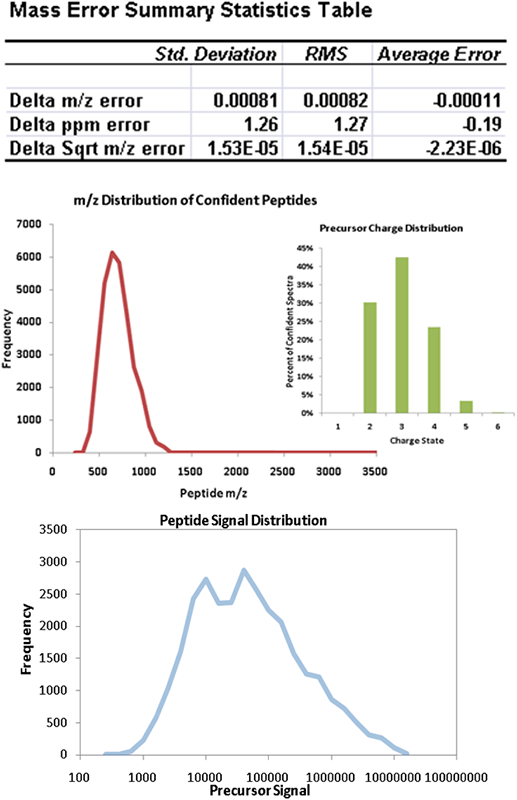
One of the keys to fully optimizing the quality of data acquired by an LC-MS system is the ability to measure the appropriate quantitative metrics on the acquisition. The ProteinPilot Report provides many helpful metrics on data quality. For MS data quality, detailed analysis of mass accuracy is performed, both overall (Figure 2, top) and as a function of retention time or precursor signal. Distributions of the charge state, mass, and m/z of confidently identified peptides are generated (Figure 2, middle). Using the precursor intensity at the peak apex, many different valuable analyses are performed, such as the precursor distribution (Figure 2, bottom) which directly measures the dynamic range of detected peptides in a sample.
Figure 2. MS acquisition characterization. Descriptive analyses are done considering only peptides identified at <5% local FDR. (Top) Table showing the analysis of mass accuracy with metrics on precision, bias, and the combined RMS metric. (Middle) Precursor mass/charge distribution intensities of peptide precursors observed in a complex cell lysate using the TripleTOF® 5600 system. (Bottom) MS intensity distribution peptide precursors showing the dynamic range of peptides identified in a human plasma sample, almost 5 orders of dynamic range.
Characterization of LC properties
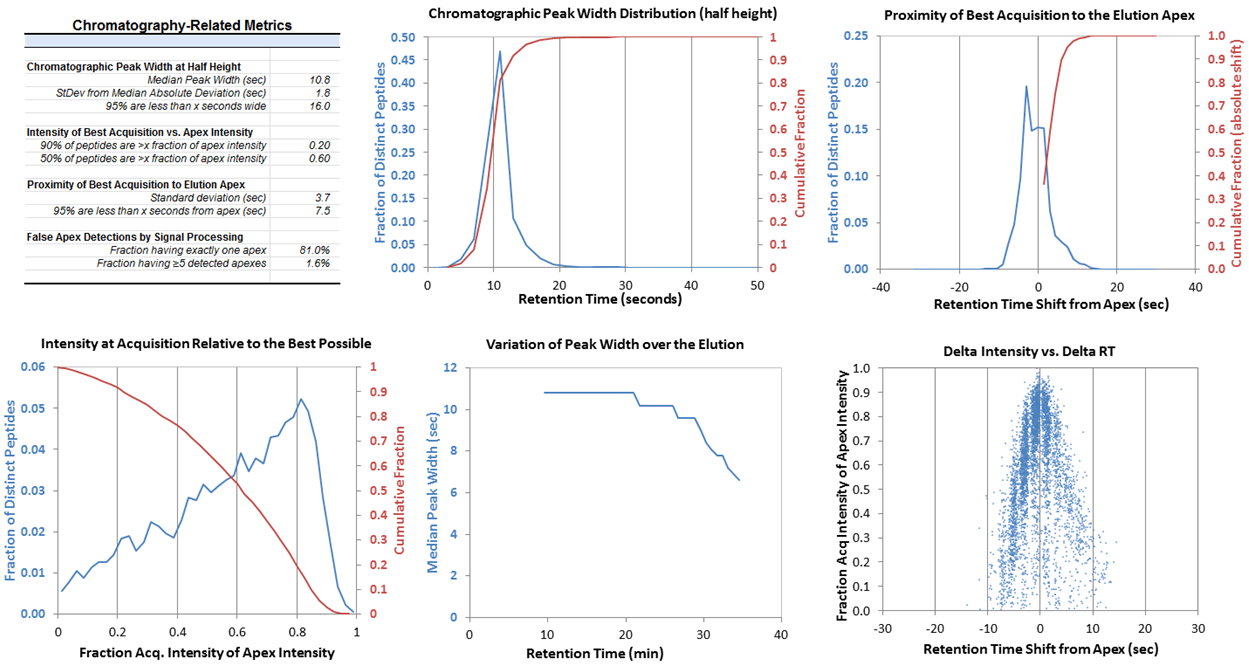
Another key aspect to high quality LC-MS analysis is the quality of the chromatography. A dashboard showing all the key properties of chromatography is available. A detailed analysis is performed on the LC peak width for each peptide and plots showing the distribution of peak widths and the median peak width as a function of retention time (Figure 3). This information can be used to assess and improve the LC separation and also during method optimization for quantitative workflows such as SWATH Acquisition or MRMHR workflow. An analysis is also performed to understand how where the MS/MS is triggered relative to the LC peak apex.
Figure 3. Chromatography characterization. Quality of chromatography is important to assess when performing any proteomics experiment. The chromatography dashboard provides a large amount of information to help understand current quality and how improvements could be made. For example, the average chromatographic peak width is very important to consider when building acquisition methods for XIC based quantitation methods, such as SWATH® Acquisition and MRMHR workflow.
Characterization of sample properties
Proteases do not have perfect cleavage specificity. Thus, the ability of the Paragon Algorithm to search for missed cleavages (under cleavage) and unexpected cleavages (over cleavage), in addition to hundreds of sample preparation and biological modifications ensures higher fidelity in the identification results. The Report provides a detailed analysis of the quality of the digestion (Figure 4). Monitoring the missed cleavage and semi-tryptic rates observed in each study is an effective way to ensure that the digestions are working well and reproducibly (Figure 4, top). The heat map (Figure 4, bottom) shows the cleavage rates observed between each residue pair for the cases where digestion did not conform to expected digestion sites.
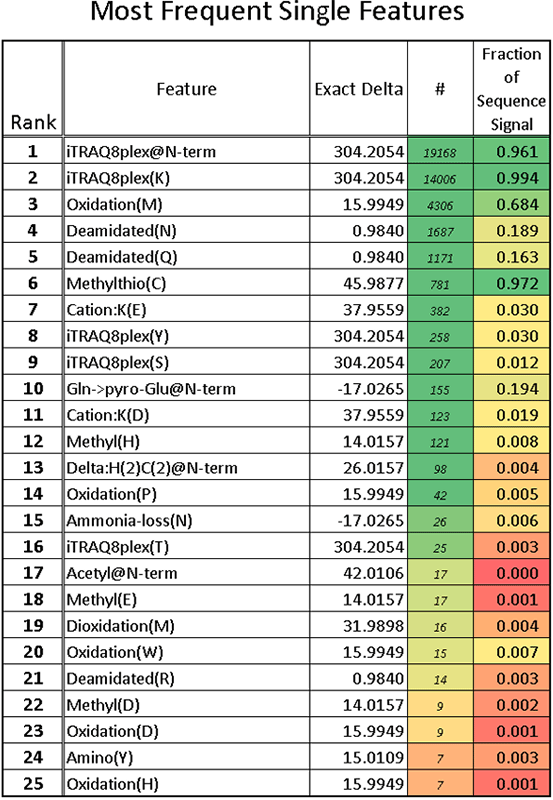
The Paragon Algorithm in Thorough mode automatically searches for 100s of sample preparation and biological modifications as well as amino acid substitutions. A detailed summary is provided as well as a distillation of the 25 most frequent modifications observed in the confidently identified peptides (Figure 5). It also computes the fraction of total ion signal having the modification of all forms of the same base sequences, as measured via peptide elution apex intensities. This allows for the rigorous QC of sample preparation steps, like cysteine alkylation, and labeling chemistries as well as undesired side reactions.
Figure 4. Characterization of digestion. Digestion frequencies are useful to monitor as deviations from normal can indicate problems with sample preparation. Shown are the observed frequencies of cleavages for a sample digested with trypsin. The frequency for each residue pair is computed as the number of observed unexpected cleavages divided by the number of possible sites (reported as frequency per 1000).
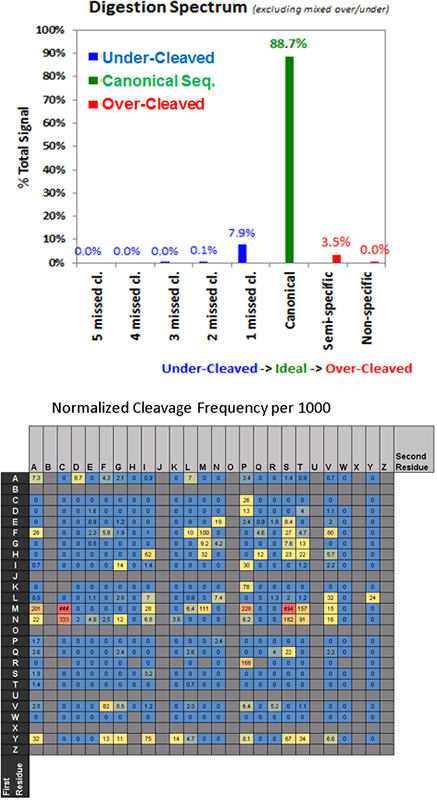
Figure 5. Summary of detected modifications. The most frequent modification table is useful for sample QC. In the example shown, it can be seen that the SCIEX iTRAQ® reagent labeling is very high (96.1% and 99.4%), as is the cysteine modification (97.2%) as measured by peptide signal.
Characterization of Quantitative Results
There are a number of dashboards that are computed to help with understanding the quality of the quantitative data obtained for the SCIEX iTRAQ® reagents or other labeling experiments analyzed. One important analysis that is done on a quantitative dataset is a target-decoy analysis of the quantitative ratios to determine the p-value cutoff to use to get a desired FDR level in the differential protein list. This can be done when there is a true analytical replicate present in the multiplex that can be used to create decoy ratios (Figure 6). Once the p-value is determined the final list of proteins can be easily pull from the tab that distills the list of differentially expressed proteins sorted by ascending p-value.
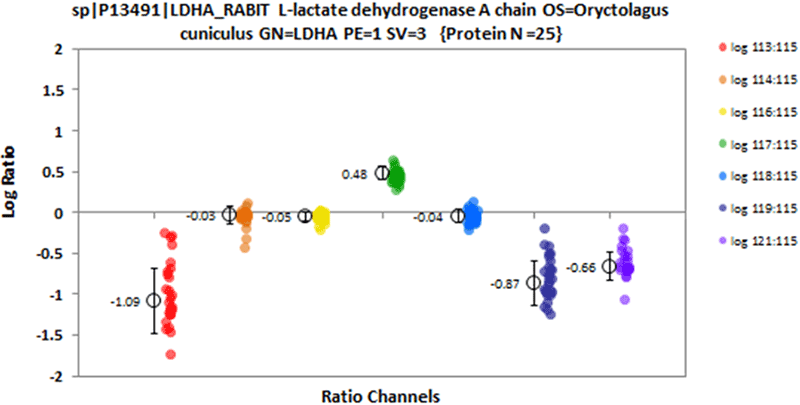
Visualization of individual protein results is possible using the protein viewer (Figure 7). Here the underlying quantitative data for specific proteins can be visualized.
Figure 6. Analysis of quantitative results using target-decoy analysis. To determine the proteins that are differentially experiments with good confidence, the Report performs a target-decoy analysis of the protein quantitative ratios. The sample design must include two channels that possess an analytical replicate such that the ratios between these two similar channels can serve as decoy ratios. The number of target vs decoy ratios are determined at each p-value level (bottom) and then this can be used to compute a FDR curve (top). A table is generated (middle) that provides a number of fixed FDR levels that one can use to choose a p-value threshold to use for the quantitative results to extract the final list of differential expressed proteins.
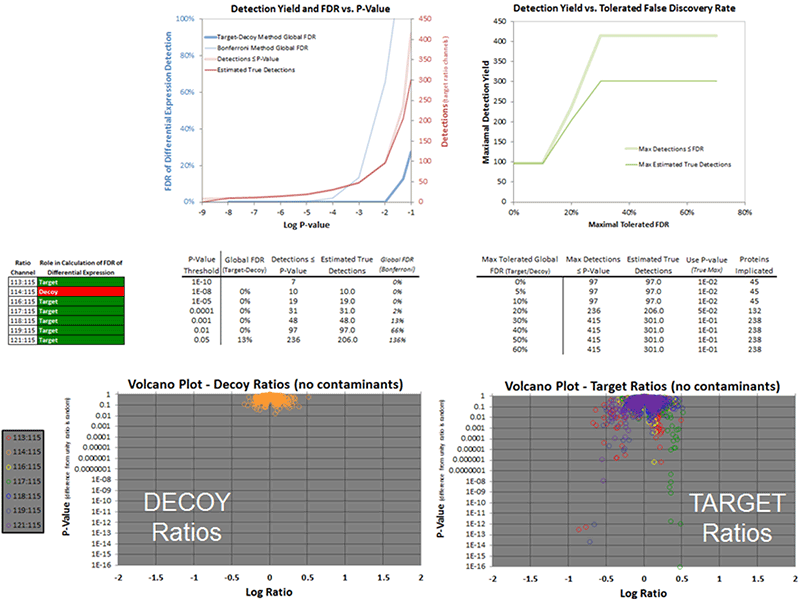
Figure 7. Single protein viewer. Once the set of differentially expressed proteins is determine, it is often desirable to visualize the quantitative ratio data for individual proteins of interest. This viewer allows for selection of a specific protein and shows the individual peptide ratio for the protein found in each of the ratio channels such that the quality of the underlying ratio data can be assessed.
Ontology analysis
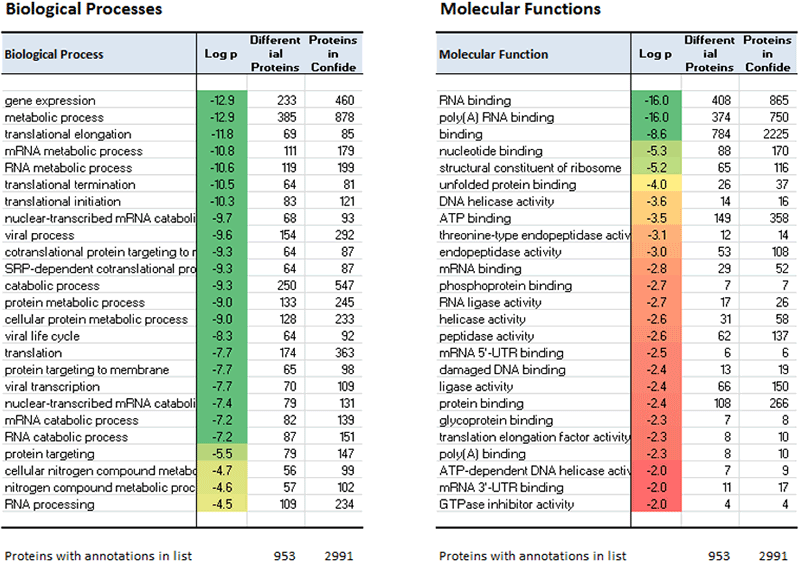
After every ProteinPilot Software database search (when searching the UniProt/SwissProt FASTA files), the UniProt website is accessed and the ontology information available for every identified protein is downloaded and incorporated into the results (*.group file). The report performs an analysis on this information and determines if there is any enrichment of any of the protein classes in the dataset or specifically in the differentially expressed proteins (Figure 8).
Figure 8. Analyzing the ontology information. Every search result is annotated with the ontologies that are available from the UniProt website, using the 10 categories of information. The ProteinPilot Report distills the information for the confidently identified proteins and performs two types of enrichment analysis: 1) Ontology distribution of proteins relative to the distribution of a reference proteome, 2) ontology distribution of differentially expressed proteins relative to identified proteins (shown above).
Conclusions
- The ProteinPilot Report is a powerful tool to provide a much deeper understanding of LC-MS identification and quantitation results, in minutes rather than weeks.
- Many detailed dashboards are provided by the Report that help characterize the quality of collected LC-MS, including sample preparation, chromatographic quality, MS acquisition quality, etc.
- Analysis of label-based quantitative experiments is provided, including metrics and graphical views of variation, volcano plots, and calculation of the false discovery rate of differential expression for some workflows.
- >7000 quantitative readouts are produced by the report, which can be captured in a single column, enabling everything from simple comparison of two data sets to complex data mining.
References
- ProteinPilot™ Software overview. SCIEX technical Note RUO-MKT-02-1777-B.
- The Paragon Algorithm, a Next Generation Search Engine That Uses Sequence Temperature Values and Feature Probabilities to Identify Peptides from Tandem Mass Spectra, Shilov IV et al. (2007), Mol. Cell. Proteomics, 6, 1638-1655.
- Nonlinear Fitting Method for Determining Local False Discovery Rates from Decoy Database Searches. Tang W et al. (2008), J. Prot. Res. 7(9), 3661–3667.
- The ProteinPilot™ reports are automatically installed and can be found at C:\Program Files\AB SCIEX\ProteinPilot\WorkflowDirectory
- How do I use the new ProteinPilot Reports (small vs large)? SCIEX community post RUO-MKT-18-8310-A.