Increasing the multiplexing of high resolution targeted peptide quantification assays
Scheduled MRMHR Workflow on the TripleTOF® Systems
Jenny Albanese, Christie Hunter
SCIEX, USA
Abstract
High resolution targeted assays can provide higher specificity for some peptides in complex biological samples. Scheduling the acquisition of high resolution MS/MS on the target peptide in a short time window around its elution time allows more accumulation time to be used per peptide while still maintaining an optimized cycle time. In this way more peptides can be monitored in a single LC-MS run with highest quantitation quality. After acquisition, extraction of fragment ions from the high resolution TOF MS/MS data allows for high specificity MRM-like data (MRMHR Workflow) to be obtained. Here, in the multiplexing test on standard protein digests, up to 800 precursors were analyzed in a single method and high reproducibility was observed. The TripleTOF Systems have the MS/MS sensitivity and speed to perform MRM-like analysis on large numbers of peptide targets in a single acquisition. This reproducibility translated into the complex biological sample test, where 130 peptides were monitored in E.coli with 90% of the transition peak areas showing reproducibility better than 20%.

Introduction
Targeted quantitative proteomics has become a key tool in proteomics research and is driven by the well-established sensitivity and selectivity attributes of multiple reaction monitoring (MRM). 1 As more extensive protein panels need to be monitored in a targeted way across multiple samples, higher MRM multiplexing is becoming essential for throughput. With the Scheduled MRM™ Algorithm (introduced with Analyst® Software 1.5), significantly more MRM transitions can be monitored in a single acquisition while maintaining the quantitative reproducibility by time scheduling the acquisition of MRM transitions. 2
The speed and sensitivity of MS/MS acquisition on the TripleTOF® Systems has enabled a high resolution targeted quantification strategy. This strategy (termed MRMHR workflow) enables a degree of selectivity using high resolution that cannot be reached using standard triple quadrupole based instruments.3
In this technical note, the Scheduled MRMHR workflow (Figure 1) that is now available in Analyst® TF Software 1.6 will be assessed for the degree of multiplexing possible and its impact on quantitative reproducibility. Scheduling the acquisition of high resolution MS/MS on the target peptide in a short time window around its elution time allows more accumulation time to be used per peptide while still maintaining an optimized cycle time. In this way more peptides can be monitored in a single LC-MS run with highest quantitation quality.
Figure 1. Scheduled MRMHR Workflow. Using knowledge of the elution time of each peptide, each precursor mass is monitored only during a short retention time window and the full scan high resolution MS/MS is acquired. This allows many more peptides to be monitored in a single LC run with higher accumulation time and an optimized cycle time for the highest quantitation quality.

Key features of Scheduled MRMHR Workflow on TripleTOF Systems
- Using the precursor mass and retention time of each peptide, large numbers of peptides can be analyzed with high resolution and high quality data in a single run
- Scheduled MRMHR workflow enables longer accumulation times to be used during acquisition to provide good analytical precision at high multiplexing
- Full scan, high resolution data from Scheduled MRMHR workflow can be searched using ProteinPilot™ Software for confirmation of detected peptides
- Scheduled MRMHR Workflow is available on all TripleTOF Systems using Analyst Software 1.6 or higher.
Methods
Sample preparation: Protein digests of Bovine Serum Albumin (Michrom BioResources), Myoglobin, (Michrom BioResources) and Beta-Galactosidase (SCIEX) were mixed together to create a test sample and loaded at 10 fmol on column. E.coli protein digest (Waters) was analyzed at 250 ng on column.
Chromatography: Separation of protein digests was performed on a nanoflow LC system using two 75 µm x 15 mm analytical columns (ChromXP™ C18-CL 3μm 120Å phase) in serial to create a longer column. Peptides were separated using a linear gradient formed by A (2% ACN, 0.1% FA) and B (98% ACN, 0.1% FA), from 12–32% of B over 60 minutes at a flow rate of 250 nL/min. Each injection was performed using a full loop injection with a 1 µL sample loop.
Mass spectrometry: The MS analysis was performed on a TripleTOF 5600 LC-MS/MS System (SCIEX) using both the MRMHR Workflow3,4 and Scheduled MRMHR Workflow. MS spectra were acquired in high resolution mode (>30,000) using 250 ms accumulation time per spectrum. Full scan MS/MS was acquired in high sensitivity mode with an accumulation time optimized per cycle. Collision energy (CE) was set using rolling collision energy with a collision energy spread (CES) of 5 V. Retention time window used for most datasets was 2.5 minutes, except for the 800 peptide method where a window of 2.0 minutes was used.
Data processing: Peptide identification on the MS/MS collected on the E.coli sample was performed using ProteinPilot™ Software and an xml file was created for import into Skyline Software (University of Washington, USA). Proteins and peptides were selected for analysis and MRMHR workflow data was acquired and processed to determine peptide retention times. Final Scheduled MRMHR workflow acquisition methods and MultiQuant™ Software quantitation methods were created simultaneously by Skyline. Final data processing was performed using MultiQuant Software.
Figure 2. Assay development process for Scheduled MRMHR Workflow. Experimental MS/MS data from IDA acquisition on TripleTOF Systems or MS/MS data from other sources can be imported for initial assay development. MRM HR workflow methods can be built for target proteins and peptides and used for assay refinement and retention time determination. Final Scheduled MRMHR workflow methods are built and used to run biological study. At the same time, a MultiQuant™ Software quantitation method is exported for use in final data processing. |
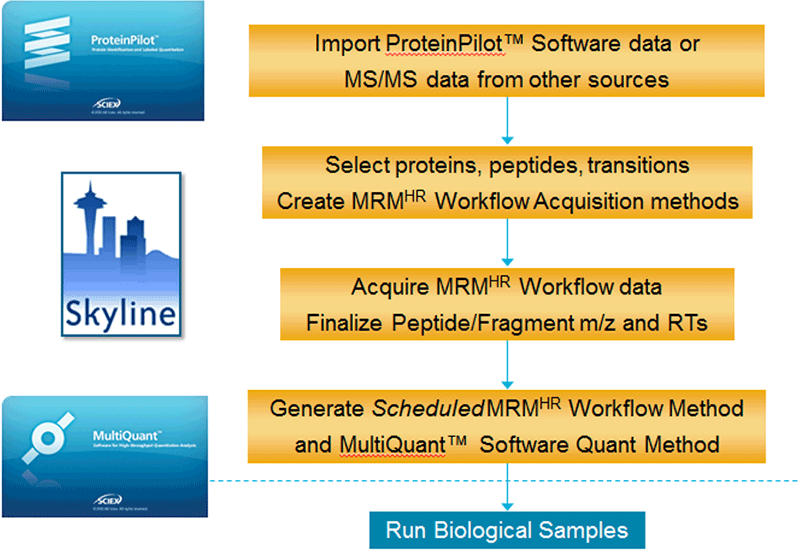
Assay development workflow
Assay development is simplified for MRMHR and Scheduled MRMHR workflow as full scan MS/MS is always acquired and fragment ion selection is done post-acquisition. Only the peptide precursor ion m/z and the collision energy are set up-front. This process of developing Scheduled MRMHR workflow assays has been implemented within the Skyline Software and consists of four simple steps (Figure 2).
- Previously acquired MS/MS data from a database search result, such as ProteinPilot Software, can be brought into the Skyline software to provide protein, peptide, and MS/MS information. Assays can also be developed from protein sequence information.
- Initial MRMHR workflow acquisition methods are constructed using the intensity information from MS/MS data
- These acquisition methods are run against the biological sample of interest and the MRMHR data is evaluated. Peptide and fragment ion selections can be refined to develop the highest quality assay (Figure 3). Retention times of each peptide are also determined
- The final Scheduled MRMHR Workflow acquisition method is built and will be used for data collection on the biological sample of interest. In addition, a MultiQuant Software quantitation method is created to be used for final data processing
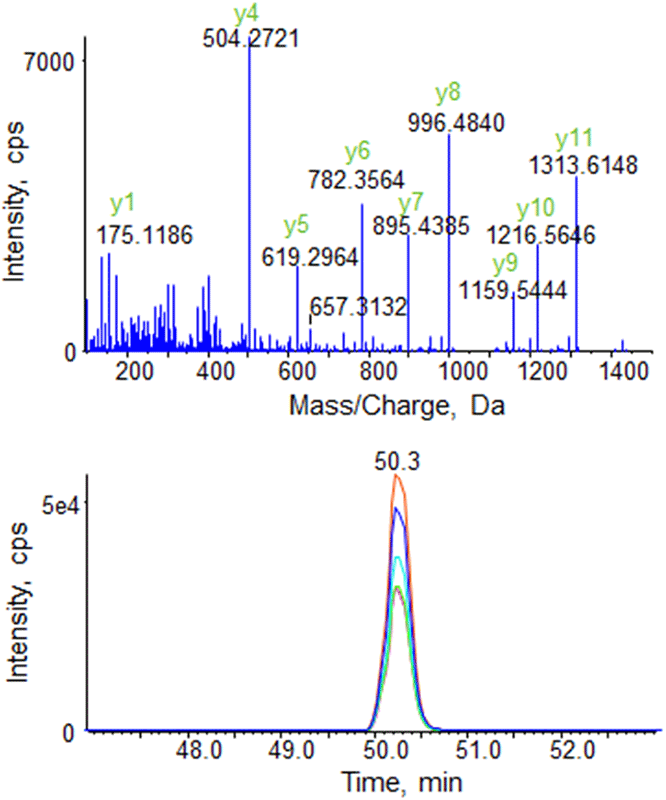
Figure 3. High resolution XIC generation from a Scheduled MRMHR Workflow demonstrates sufficient points across an LC peak. To quantify the peptide DGEDPGYTLYDLSER (m/z 577.2566) from Beta-gluatmate decarboxylase in E.coli cell lysate, a looped MS/MS spectrum is acquired across the LC peak at high resolution (>15,000) (top). High resolution XICs are generated for the peptide post-acquisition (bottom). Scheduling the acquisition of the MS/MS spectra around the elution time for the target peptide enables higher multiplexing while maintaining data quality.
Assessing quantitative reproducibility at high multiplexing
An experiment was designed to assess the effects of higher multiplexing on analytical reproducibility on the TripleTOF 5600 system. Thirty peptides from Beta-Galactosidase (BG), Horse Myoglobin (Mb) and Bovine Serum Albumin (BSA) protein digests were monitored using the MRMHR Workflow with a cycle time of 1.8 seconds. Random generated Q1 m/z values and retention times were then added to the real peptides and these were used to build Scheduled MRMHR workflow methods with the same cycle time of 1.8 seconds and a retention time window of 2.5 minutes. Each acquisition method was run on the protein digests in 5 or more replicates by NanoLC. The reproducibility of fragment ion XICs for the 30 real peptides from each method across the replicate injections was used as a measure of the analytical reproducibility (Figure 4). The total number of XICs (in %) at a specific % coefficient of variation (%CV) was computed and plotted in Figure 4 (top), the median %CV was also computed (Figure 4, bottom). This cumulative reproducibility plot shows the majority of XICs achieved %CV of less than 20%, even at the highest peptide multiplexing in this experiment. Scheduled MRMHR workflow for 200 and 400 peptides (orange and blue) showed reproducibility similar or better to the MRMHR workflow data (green) on 30 peptides as the MS/MS accumulation time was greater than 40 msec in those methods. For 600 peptides (red line), there was a small drop in data quality as the accumulation time decreased to 30 msec. To run 800 peptides (purple line), the retention time window was reduced from 2.5 minutes to 2 minutes to maintain an accumulation time of around 30 msec and good quantitative reproducibility was observed.
In addition, the effect of multiplexing on quantitative reproducibility for a set of peptides from proteins across the abundance range in an E.coli digest was studied. A total number of 139 peptides from 32 proteins were analyzed in a single LC/MS run using Scheduled MRMHR workflow and 795 fragment ions were quantified. Replicate analysis was performed and the reproducibility was assessed at both the transition XIC area and peptide level (multiple fragment ion XIC areas were summed to obtain a peptide area). Figure 5 shows extremely low variance at the transition XIC level (blue trace) and across replicates with 90% of data having CVs of less than 20% and over 95% of data at the peptide level (orange trace). The reproducibility of the retention times for the peptides across the replicate injections was an average of 0.2% RSD.
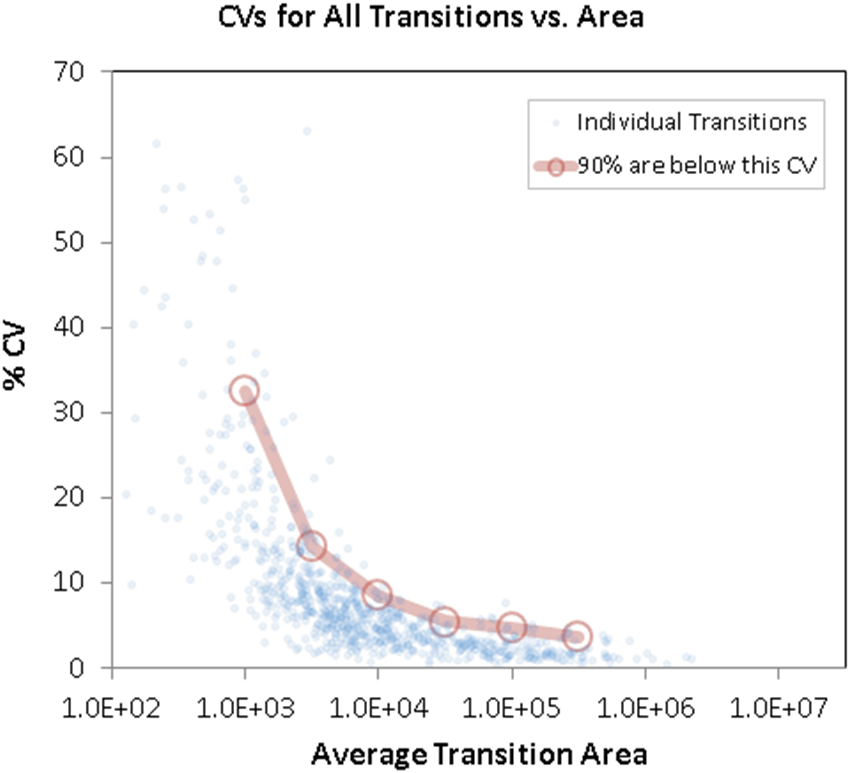
To evaluate the XIC peak area quality and reproducibility further, each individual data point was plotted based on its intensity and %CV (Figure 6). The variance in the data increases at the lower intensity regimes as expected. The red line shows that only at the very lowest intensity bins does the %CV exceed 10%.
Figure 4. Assessing the quantitative accuracy of higher multiplexing using Scheduled MRMHR Workflow. The effect of higher numbers of peptides on the reproducibility of 30 peptides across 5 or more replicate injections on the TripleTOF 5600 system was assessed. Because the data was acquired using the Scheduled MRMHR Workflow, the reproducibility of up to 800 peptides showed very good quantitative accuracy.
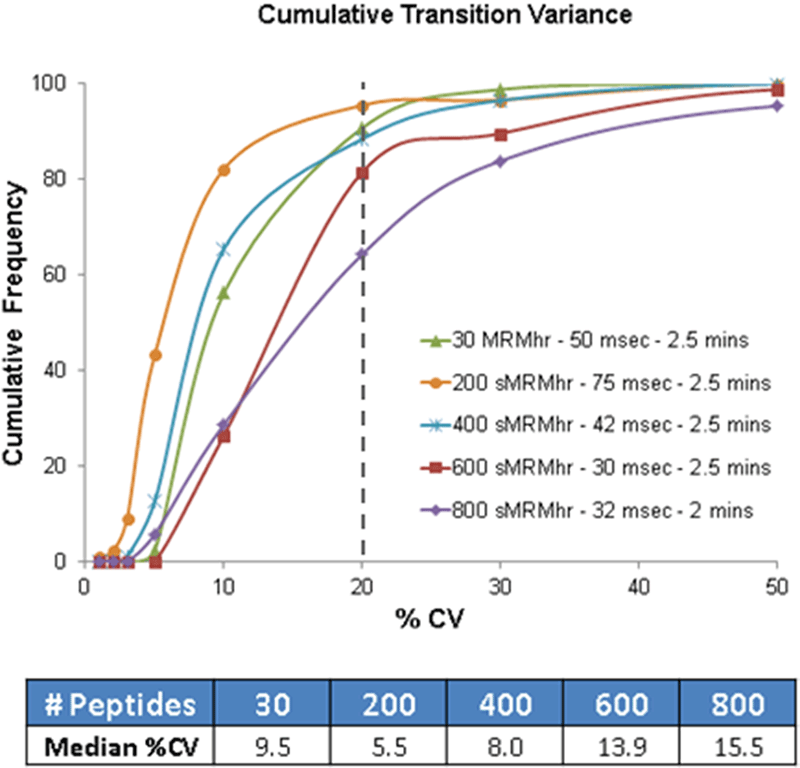
Figure 5. Quantitative reproducibility for peptides in E.coli cell lysate digest. 139 peptides from proteins across the abundance range in an E.coli digest were monitored using the Scheduled MRMHR workflow. Replicate analysis was performed and the reproducibility at both the XIC and peptide level was assessed. Extremely low variance was found at the transition XIC level (blue trace) across replicates with 90% of data having CVs of less than 20% and over 95% of data at the peptide level (orange trace). |
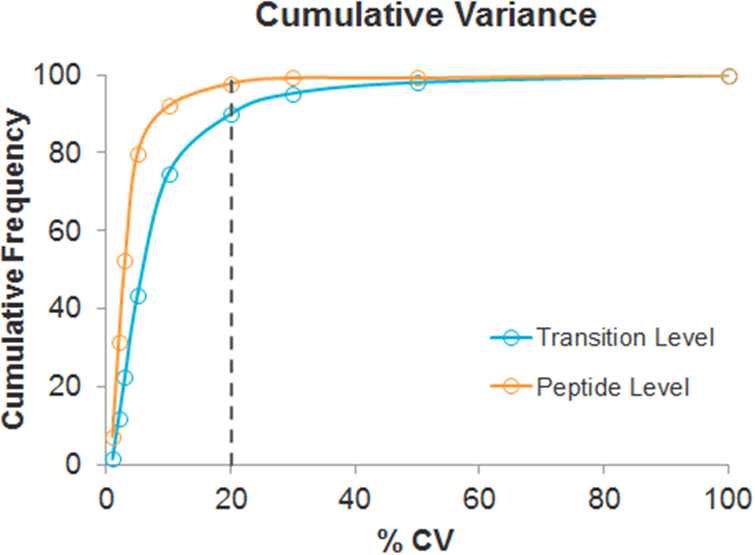
Figure 6. Reproducibility assessment of transition peak area versus %CV. The blue dots represent the individual transition peak areas and are plotted based on their intensity and observed variance. The red line describes that 90% out of the total transitions area are below the shown %CV. This demonstrates the very high data reproducibility that was obtained by using the Scheduled MRMHR workflow on the TripleTOF 5600 system. |
Increased specificity on a TripleTOF Systems
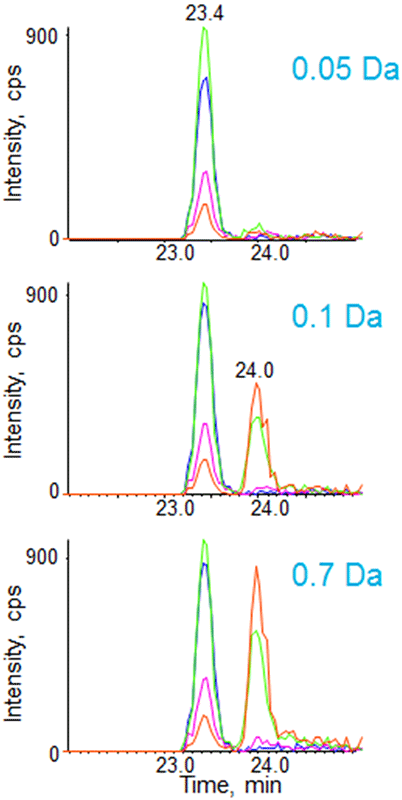
When targeted quantification is done on a triple quadrupole based instrument, quadrupoles are typically set at unit resolution, transmitting a 0.7 Da wide window in both Q1 and Q3. The difference with the MRMHR Workflow is that the TOF analyzer replaces the ‘Q3’ fragment ion detection with its inherent advantages of detecting all ions, at high resolution and high mass accuracy. To illustrate the advantage of high resolution, multiple fragment ions for the peptide VNAIAAPTR from an uncharacterized protein yraR were extracted at variable extraction widths (Figure 7). At the narrow extraction widths possible from the high resolution TOF data, good specificity is observed with minimal interfering peaks (Figure 7, top). When the extraction width is widened to simulate monitoring using a quadrupole, an interference is observed for two of the transitions (Figure 7, bottom).
Figure 7. Removing interferences or background through high resolution fragment ion extractions. Multiple fragment ions for the peptide VNAIAAPTR from an uncharacterized protein yraR were extracted at variable extraction widths. High resolution extraction of fragment ions can provide higher specificity in complex matrices when specific interferences are present. |
Conclusions
High resolution targeted assays can provide higher specificity for some peptides in complex biological samples and now with the ability to time-scheduled acquisition, high multiplexing can be achieved.
- TripleTOF Systems have the MS/MS sensitivity and speed to perform MRM-like analysis
- Post-acquisition extraction of fragment ions from the high resolution TOF MS/MS data allows for high specificity MRM-like data (MRMHR Workflow) to be obtained
- Scheduled MRMHR Workflow extends the multiplexing possible with this high resolution workflow, maintaining quantitative robustness
In the multiplexing test on standard protein digests, up to 800 precursors were analyzed in a single method and high reproducibility was observed
This reproducibility translated into the complex biological sample test, where 130 peptides were monitored in E.coli with 90% of the transition peak areas showing reproducibility better than 20%
References
- Anderson L and Hunter CL (2006) Mol. Cell. Prot. 5.4, 573-588.
- The Scheduled MRM™ Algorithm Pro - Automated intelligent design of high throughput, high quality quantitation assays. SCIEX technical note RUO-MKT-02-8539-A.
- Impact of high-resolution MRM analysis on complex proteomes- Targeted protein quantification using MRMHR Workflow on the TripleTOF® 5600 System. SCIEX technical note RUO-MKT-02-5733-A.