Large-scale protein identification using microflow chromatography on the ZenoTOF 7600 system
Cloud-based processing with the ProteinPilot application in OneOmics suite
Alexandra Antonoplis1, Nick Morrice2 and Christie Hunter1
1SCIEX, USA, 2SCIEX, UK
Abstract
Generation of spectral libraries is often thought of as a time consuming procedure, but in reality they can be generated very quickly. Using microflow chromatography with very fast MS/MS acquisition on a QTOF system, then searching the data in the cloud can enable library generation in <48 hrs. Here this workflow was explored using the ZenoTOF 7600 system, and the impact of larger libraries on DIA protein quantification was tested. The libraries generated from each fractionated cell line contained >8000 proteins and >175000 peptides.

Introduction
Data-dependent acquisition (DDA), also known as information-dependent acquisition (IDA), remains an important workflow for the proteomics researcher to determine the protein content of a sample. It is also key in the generation of spectral libraries for data-independent analysis (DIA), a powerful acquisition mode for targeted extraction and comprehensive quantification of peptides and proteins.1 Deep interrogation of a digested proteome sample or generation of a large DIA library can be time-consuming, but a recent strategy involving extensive fractionation of cell digest followed by fast microflow LC-MS analysis and cloud-based DDA data processing enabled library generation in less than a day.2
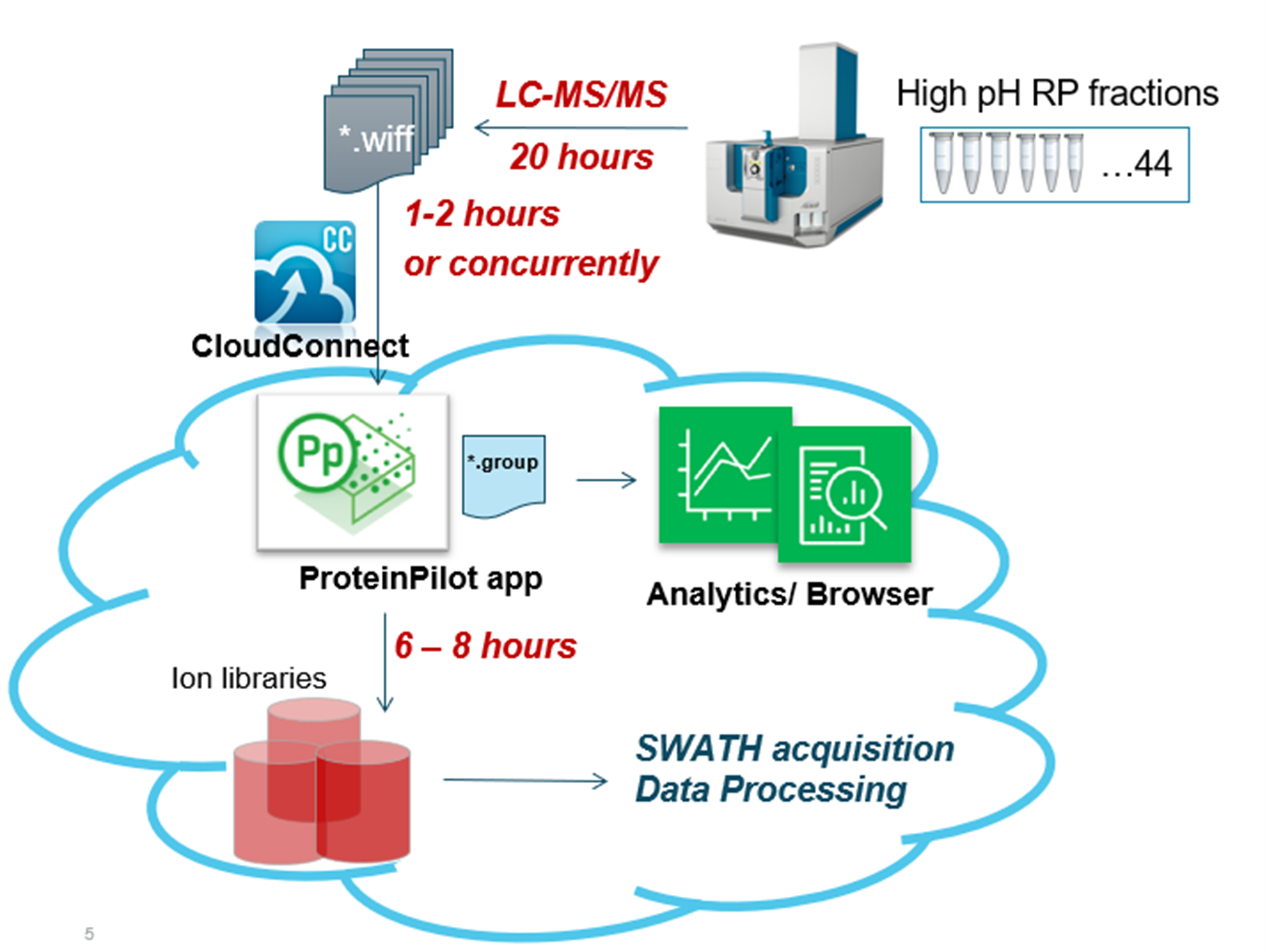
In this work, this strategy was further improved using the ZenoTOF 7600 system, a QTOF platform equipped with Zeno MS/MS for enhanced sensitivity. Zeno trap activation on this system has been shown to provide 5- to 20-fold gains in MS/MS sensitivity and up to 40% gains in protein identifications.3,4 Using this MS platform in combination with fractionated cell digest samples and fast microflow chromatography, ion libraries were generated for further use in SWATH acquisition proteomics workflows (Figure 1).
Figure 1. Overview of the protein identification and library generation workflow. Forty-four high-pH fractions were analyzed by microflow chromatography and data-dependent acquisition with Zeno MS/MS on the ZenoTOF 7600 system. CloudConnect was used to upload data files to OneOmics suite for processing in the ProteinPilot application. The resulting libraries were then used to process SWATH acquisition data.
Key features of the library generation strategy using the ZenoTOF 7600 system
- Forty-four high-pH fractions were prepared for each proteome digest· Full workflow, from fractionated samples to deep proteome interrogation for spectral library generation, took less than 48 hours (Figure 1)
- Fast, 21-minute microflow LC gradients were used to rapidly analyze the peptide fractions
- The ZenoTOF 7600 system was used with Zeno IDA to generate high-quality MS/MS data with ~5-6x higher sensitivity3,4 and at least 2x more sequence coverage relative to Zeno trap off
- Data were uploaded to OneOmics suite and searched using the ProteinPilot application to identify many proteins from the fractions and generate an ion library for processing SWATH acquisition data.
Methods
Sample preparation: A 100 µg sample of digested HeLa or K562 cell lysate was fractionated using high-pH RP-HPLC. For high-pH RP-HPLC, Buffer A was 2mM ammonium hydroxide in 2:98 (v/v) acetonitrile/water and buffer B was 2mM ammonium hydroxide in 90:10 (v/v) acetonitrile/water. An ExionLC system with a UV detector was used for fractionation with a flow rate of 1 mL/min and a 250 x 4.6 mm Durashell RP column. In total, 44 fractions were manually collected and then dried. Samples were resuspended in 40 µL of 2:98 (v/v) acetonitrile/water with 0.1% formic acid, then 10% of each fraction was injected for DDA. For SWATH acquisition, a sample of K562 digest (SWATH acquisition performance kit) was prepared in water with 1% formic acid and analyzed at varying loading amounts.
Chromatography: The Waters ACQUITY UPLC M-class system was used for all separations with a Phenomenex Kinetex 2.6 µm XB-C18 LC column (100 Å, 150 x 0.3 mm). Mobile phase A was water with 0.1% formic acid and mobile phase B was acetonitrile with 0.1% formic acid. For analysis of the HeLa and K562 fractions, a rapid, 21-minute linear gradient from 5 to 30% mobile phase B was used with a flow rate of 6 µL/min in direct inject mode. For SWATH acquisition experiments, a 45-minute microflow gradient from 5 to 30% mobile phase B was implemented in trap-elute mode with a Phenomenex C18 micro trap (10 x 0.3 mm) and a flow rate of 5 µL/min.
Mass spectrometry: A ZenoTOF 7600 system equipped with the OptiFlow Turbo V source using a low microflow probe and electrode was used for all data acquisition. DDA parameters that were implemented for all experiments included a TOF-MS accumulation time of 100 ms, an exclusion time of 5 seconds and activation of the Zeno trap. Only precursors with charge states ranging from 2 to 4 with intensities greater than 300 cps were selected for fragmentation. The maximum number of candidate ions per cycle was 45 and the accumulation time was 20 ms. For SWATH acquisition, a 80 variable window method was used with an MS/MS accumulation time of 25 ms and dynamic collision energy.
Data processing: Data files were uploaded to the SCIEX Cloud platform using CloudConnect in PeakView software v2.2. Data were then searched using the multi-file option in the ProteinPilot application in OneOmics suite using a UniProt human FASTA file. The “thorough” search effort was used, and the “biological modifications” option was selected to ensure the broadest search space. Search results were visualized using the Analytics and Browser applications to ensure the quality of the protein identification results. Data from each fractionation experiment in each cell line were processed separately in the ProteinPilot application. Processed data were then merged using the Extractor application to create a final ion library. The generated ion libraries were then used to process SWATH acquisition data in DIA-NN software. In DIA-NN software, the robust LC, high precision workflow setting was selected with MBR implemented and all other settings set to software defaults.
Protein identification in the cloud-based ProteinPilot application
Zeno IDA data were collected for all K562 and HeLa digest fractions and processed using the cloud-based ProteinPilot application in OneOmics suite. The ProteinPilot application provides several visuals to assess data quality, including mass accuracy tables, digestion metrics and dynamic range plots. For the ion libraries, high-quality digestion and high mass accuracy in the underlying data were observed, illustrating the quality of the library generation approach (Figure 2). The peak areas of the detected peptide precursors were also plotted, highlighting more than 5 orders of dynamic range in the dataset.
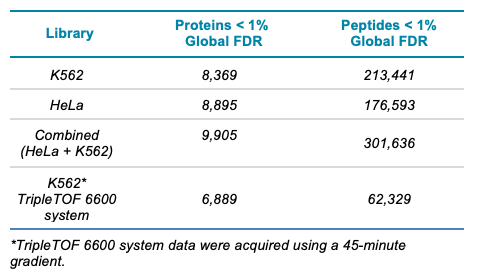
The data were then interrogated for overall protein and peptide identifications using the cutoff of <1% global FDR. The final K562 digest library contained 8,369 proteins at <1% global FDR and 213,441 peptides at <1% global FDR (Table 1). Compared to a previous library generation approach implemented using the TripleTOF 6600 system in which 20 fractions of K562 digest were run with 45-minute microflow gradients2, 1.2x more proteins and 3.4x more peptides were observed using the current approach (Table 1). Combining both the HeLa and K562 data resulted in a library with 1,000 more proteins than each of the individual libraries. This combined library was then used in subsequent SWATH acquisition data processing to examine its performance.
Figure 2. Examples of visuals provided in the ProteinPilot application for assessing data quality. (Top) Precursor mass accuracy for K562 digest library. A table of summary statistics is provided to illustrate mass error. (Middle) The proportion of under- and over-cleaved peptides, illustrating the quality of digestion in the K562 library. (Bottom) The dynamic range in peptide precursor areas spanned more than 5 orders.
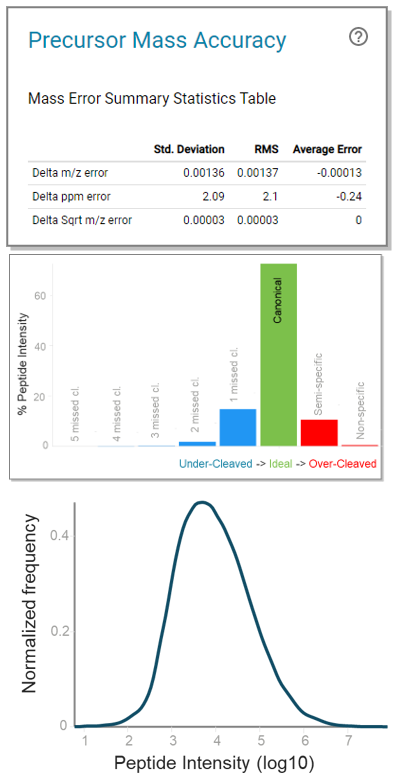
Table 1. Proteins identified using fractionation then LC-MS/MS on the ZenoTOF 7600 system. A comparison to a previous K562 library generated using the TripleTOF 6600 system is provided for reference.6
Performance of the Zeno IDA library for analysis of SWATH acquisition data
The combined K562-HeLa library generated with Zeno IDA was compared to the Pan-Human Library in a SWATH acquisition data processing workflow to assess its performance. The Pan-Human Library contains over 10,000 proteins and was generated from many different proteome DDA datasets collected on previous generation instruments over a span of several months.7 A SWATH acquisition dataset collected on the ZenoTOF 7600 system from varying K562 digest loads using a 45-minute gradient was processed in DIA-NN software using each library. The combined K562-HeLa library yielded similar performance to the Pan-Human Library (Figure 3) but was created in significantly less time, requiring less than 48 hours to generate. Comparable performance in the numbers of proteins identified and quantified was observed across several loads of K562 digest. At the peptide level, more peptides were identified with the PHL library at higher loads (100-300 ng), but this increase in peptides did not result in an increase in proteins identified. These results suggest that Zeno IDA on the ZenoTOF 7600 system enables the rapid generation of high-quality spectral libraries for use in SWATH acquisition data processing workflows.
Figure 3a. Comparison of the combined K562-HeLa library performance relative to the Pan-Human Library. SWATH acquisition data collected using a 45-minute microflow gradient and 5 µL/min flow rate were processed in DIA-NN software using matched processing settings and the 2 spectral libraries. The combined K562-HeLa library generated using Zeno IDA exhibited similar performance to the Pan-Human Library in terms of the number of proteins identified at 1% FDR and quantified at 20% CV across a variety of loads, including 13, 25, 50, 100, 200 and 300 ng.
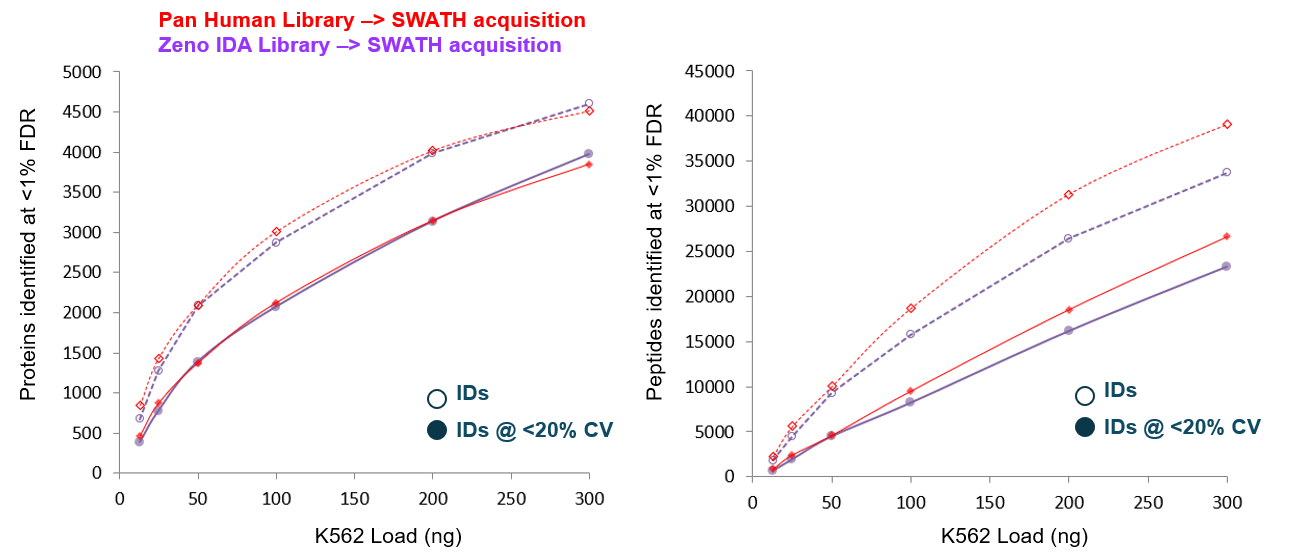
Figure 3b. Comparison of the combined K562-HeLa library performance relative to the Pan-Human Library. The Venn diagram displays protein identifications at a 300 ng load with the Pan Human (left) and Zeno IDA library (right).

Conclusions
Large-scale protein identification results from both HeLa and K562 digests were generated using high-pH fractionation followed by fast microflow analysis using the ZenoTOF 7600 system. The entire acquisition workflow from fractionated samples to spectral libraries took less than 48 hours, incorporating robust microflow chromatography and Zeno IDA. Cloud-based data processing in the ProteinPilot application enabled streamlined processing of results and assessment of data quality. The combined HeLa and K562 protein identification results were then used to build a large ion library. This Zeno IDA library provided comparable results to the previously established Pan-Human Library for processing of SWATH acquisition data.
References
- Improved data quality using variable Q1 window widths in SWATH acquisition. SCIEX technical note, RUO-MKT-02-2879-B.
- Spectral library generation for SWATH acquisition in less than 20 hrs. SCIEX technical note, RUO-MKT-02-12767-A.
- Qualitative flexibility combined with quantitative power. SCIEX technical note, RUO-MKT-02-13053-A.
- Over 40% more proteins identified using Zeno MS/MS, SCIEX technical note, RUO-MKT-02-13286-B.
- Large-scale, targeted, peptide quantification of 804 peptides with high reproducibility, using Zeno MS/MS. SCIEX technical note, RUO-MKT-02-13346-A.
- Extending depth of coverage with SWATH acquisition using deeper ion libraries. SCIEX technical note, RUO-MKT-02-3247-A.
- Rosenberger G et al. (2014) A repository of assays to quantify 10,000 human proteins by SWATH-MS. Scientific Data, 1, 140031.
Related content
- Zeno MS/MS significantly improves quantification for iTRAQ reagent-labeled proteomic samples. SCIEX technical note, RUO-MKT-02-14274-B.