Extending depth of coverage with SWATH® Acquisition using deeper ion libraries
Data independent acquisition on TripleTOF® 6600 Systems
Joerg Dojahn1, Nick Morrice2, Christie Hunter3
1SCIEX, Germany, 2SCIEX, UK, 3SCIEX, USA
Abstract
The SWATH® Acquisition data independent acquisition strategy provides a comprehensive quantitative analysis of complex samples, providing full scan high resolution MS and MS/MS on all detectable compounds eluting off the column. These datasets can then be re-interrogated over and over again as new information on new compounds of interest emerges. Specificity can be optimized by balancing the Q1 window width, the number of Q1 windows and accumulation time per window, such that analyte S/N can be improved significantly while maintaining good quality quantitative results. Optimization to scale the window size with precursor density, based on the matrix being analyzed, can further improve results.

Introduction
Previous work has demonstrated that the depth of coverage of SWATH Acquisition can be extended through acquisition optimizations, aimed at improving the signal/noise ratio of the quantitative data. The extended dynamic range of the TripleTOF 6600 System has enabled improved quantification across the broad concentration range of analytes found in biological samples. Fast MS/MS acquisition speeds have been leveraged to enable the use of 100 Q1 isolation windows, decreasing the complexity of data within each MS/MS window and thereby improving the specificity. Combined with variable window acquisition, very narrow windows can be used in precursor dense portions of the peptide m/z range, while still maintaining broad m/z coverage.1
A less explored variable on depth of coverage is the impact of the size of the ion library. Previous studies have suggested that more quantitative data is available in a SWATH Acquisition data files than is accessed from a small ion library generated from 1D data dependent acquisition experiments (Figure 1). Here, the impact of using larger and deeper ion libraries on the numbers of peptides and proteins detected was explored and the quality of quantitation observed by processing the same data file with increasingly deeper ion libraries was assessed.
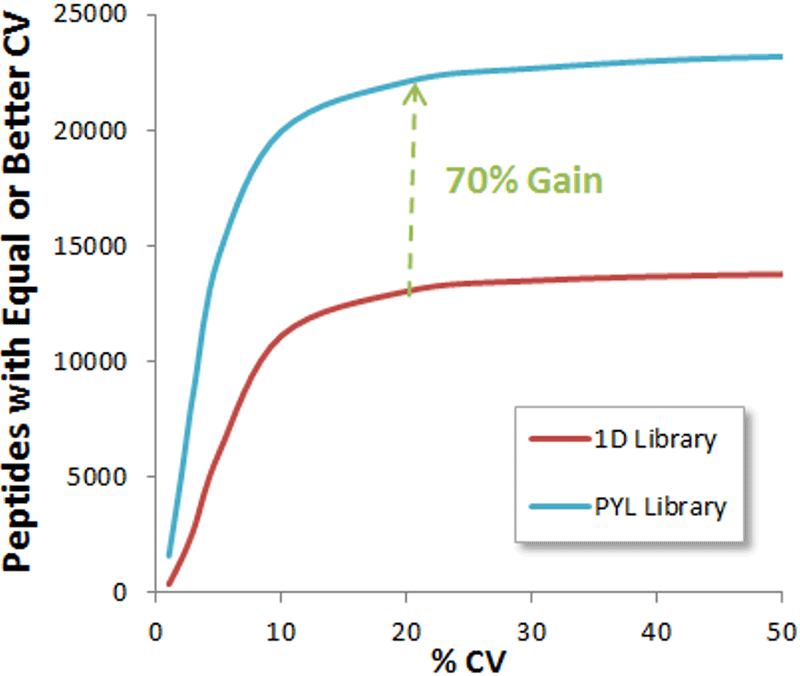
Figure 1. Gain in peptides quantified in yeast using a deeper ion library. SWATH Acquisition was collected on a yeast cell lysate using a 2 hour gradient and 3 µg total protein and using a 100 variable window method1. The data was analyzed using both a 1D ion library as well as the Pan Yeast library4 and the numbers of peptides quantified was assessed. Using a 1% FDR filter at the peptide level and computing the %CV for 5 replicate injections, the cumulative %CV curves can be plotted. Using a 20% CV as a cutoff, a gain of 70% quantified peptides was observed when using the larger Pan Yeast library.
Key features of SWATH Acquisition for quantitative proteomics
- High sensitivity, resolution and speed of MS/MS acquisition on the TripleTOF Systems
- Enables the use of 100 Q1 window isolation for improved data quality through increased specificity
- Variable sized Q1 windows optimized based on precursor density further increases specificity while ensuring broad mass range coverage2.
- Targeted data processing using large spectral ion libraries with specific fragment ions, for detection confirmation and quantitation of large numbers of peptides and proteins
Methods
2D LC-MS/MS fractionation: A digest of a K562 human cell lysate (Promega) was fractionated using high pH reverse phase chromatography using a Shimadzu Nexera system (UV detection at 214nm). A Durashell RP column (250 x 4.6mm, 5um, Agela Technologies) operating at 1 mL/min was used, running a gradient of 2 – 90% acetonitrile in 2mM ammonium hydroxide. 15 fractions were collected every 2 mins.
Each subsequent peptide fraction was then separated using low pH reverse phase gradient on the NanoLC™ 425 system operating in microflow mode. A Triart C18 150 x 0.3mm column (YMC) was used at 5 µL/min flow rate with a 45 min gradient from 2-40% acetonitrile in 0.1% formic acid. The eluent was analyzed using the TripleTOF 6600 System equipped with a DuoSpray™ Source and a 25 µm ID electrode. Data dependent acquisition was performed with 30 MS/MS per cycle, each with 50 msec accumulation.
1D LC-MS/MS chromatography: Separation of a trypsin digest of HEK human cell lysate or yeast cell lysate was performed on a NanoLC™ 425 System. A 75 µm x 30 cm C18 nanocolumn was used with a long gradient (5-30% acetonitrile, 0.1% formic acid in water) at 300 nL/min. Gradient lengths for human analyses were 60 mins for 1D library generation and 180 mins for SWATH Acquisition. For yeast, the gradient length for both 1D library generation and for SWATH Acquisition was 120 mins.
Mass spectrometry: The MS analysis was performed on a TripleTOF 6600 System (SCIEX) using a NanoSpray® Source (SCIEX). Information dependent acquisition was acquired on the fractionated and unfractionated lysates, using the 30 MS/MS per cycle with 50 msec accumulation time. Variable window SWATH Acquisition method was built using Analyst® TF Software 1.7 using 100 variable windows and 25msec accumulation times.
Data processing: 1D and 2D libraries were created by processing the IDA data using ProteinPilot™ Software. SWATH Acquisition data was processed using SWATH 2.0 Micro App in PeakView® Software 2.2 using the various ion libraries excluding modified and shared peptides. Results analysis was performed in Excel using the SWATH Acquisition Replicates template. All protein and peptide numbers reported were determined at <1%FDR and <20% CV across the 5 replicates collected.
Table 1. SWATH Acquisition ion libraries for human. A number of ion libraries of increasing size were created for interrogation of SWATH Acquisition data (at 1% FDR, excluding modified and shared peptides).
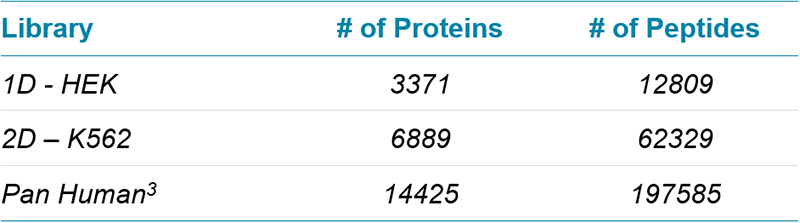
Table 2. SWATH Acquisition ion libraries for yeast. Two ion libraries of increasing size were created for interrogation of SWATH Acquisition data (at 1% FDR, excluding modified and shared peptides).
Creating ion libraries for SWATH Acquisition
An ion library for targeted data processing of data independent acquisition data contains information that characterizes the peptides for many proteins in a proteome. The masses of the peptide ion and its sequence specific fragment ions, the relative fragment intensity information, and the relative retention time information are the key information pieces and are easily obtained by performing a simple data dependent acquisition and database search. However, it is worth taking the extra time to create a deeper ion library for the biological system under study as much more information can be extracted from SWATH Acquisition data (Figure 1).
A 2D fractionation followed by LC-MS/MS protein identification on a specific complex sample can be performed in a matter of days using microflow LC which generates much deeper coverage of the proteome sample. This is well worth doing for a sample that will be studied repeatedly by SWATH Acquisition.
Some groups have invested significantly more time in library generating using multiple cell types and a large degree of fractionation to cover as much of the proteome as possible.2,3 These libraries have high value and are easily shared between research groups.
Deeper quantification in human cell lysates
As shown in Figure 1, the detection of peptides from a yeast cell lysate can be greatly enhanced by using a much deeper ion library; in this case a library generated with a 1D protein identification (Table 2) was compared with using a pan yeast library4.
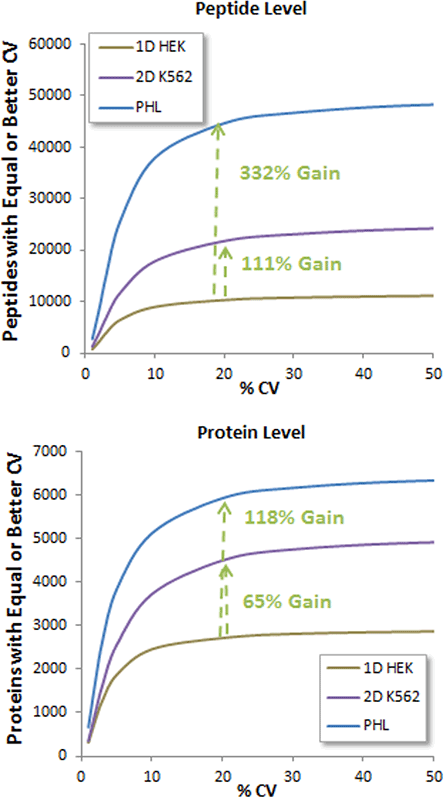
The SWATH Acquisition data files used here for a more detailed investigation was five replicates of a 3 hour nanoflow gradient run on a 3 µg load of a HEK human cell lysate on the TripleTOF 6600 System. Figure 2 summarizes the data processing experiments run on this dataset with 3 different libraries.
- The 1D HEK library was generated using 3 IDA replicates (60min gradient of 2 µg protein) run on the same cell lysate sample. This generated a small library (Table 1) of 3371 proteins and 12809 peptides. Using this library for processing, 2714 proteins and 10310 peptides were quantified with <1% FDR peptides and <20% CV (Figure 2).
- Another library (2D library on K562 cell line) was generated using a more in-depth strategy, by performing off-line fractionation on the cell line and performing protein identification using IDA on all the fractions. This larger library (Table 1) yielded a 111% gain in peptides and a 65% gain in quantified proteins (4493 proteins and 21778 peptides total) from the same SWATH Acquisition data (Figure 2).
- Finally, a pan human library has been generated using a large number of different human cell lines and extensive protein identification experiments4. When this library is used to process the same SWATH Acquisition replicate data, another significant gain in quantified peptides and proteins (332% and 118%, respectively) was observed over that of the 1D library experiment (Figure 2) with 5922 proteins and 44634 peptides quantified at <20% CV.
Figure 2. Impact of deeper ion libraries on extraction of quantitative data from human cell lysate SWATH Acquisition data. The same set of SWATH Acquisition replicates from a HEK cell lysate were processed with increasingly larger libraries and the proteins/peptides quantified with <1% FDR peptides and CVs were plotted. Significant gains in both peptides and proteins quantified at <20% CV were observed as the size of the ion library used for targeted data processing increased. |
In total, a 332% gain in peptides and 118% gain in quantified proteins was observed between using a simple ion library and an extensive ion library (Figure 2). This highlights the significantly deeper information that is contained in the SWATH Acquisition data over traditional DDA data. The quality of quantitation is key aspect to consider when to assessing improvements in data processing strategies. Even as more data is extracted from the data files using the deeper libraries, the quality of quantitation is maintained even into the low abundant protein/peptide regime (Figure 3).
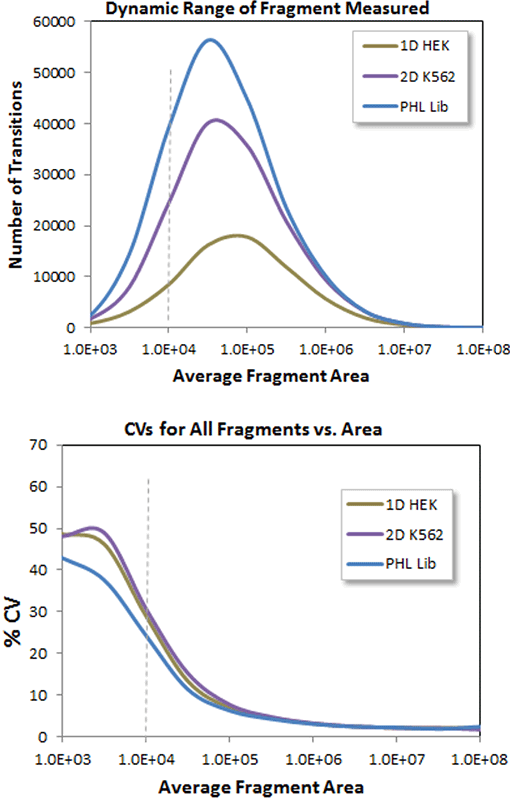
Figure 3. Dynamic range and reproducibility of the increased quantification with deeper libraries. As more peptides are quantified at lower and lower abundance (top) due to the use of increasingly larger ion libraries, it is important to monitor variance in quantitation (%CV across 5 replicates). Here the bulk of fragment ions quantified are above a peak area of 1e4 (top) and the CV vs intensity plot (bottom) shows that the bulk of the fragment ions have CVs in the 10-20% CV range.
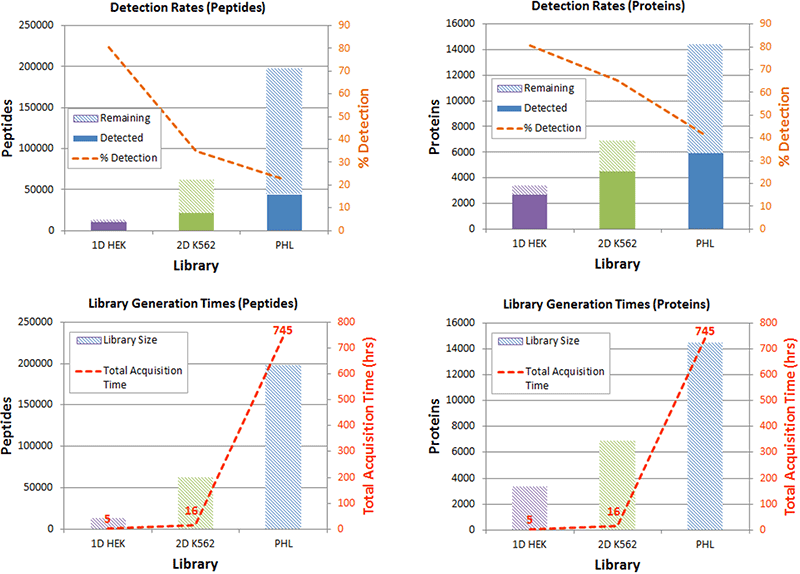
Depending on the depth of the ion library needed and the available time for analysis, a researcher can balance the library generation effort required for a particular biological system. The detection rates for each library were computed, comparing quantified peptides/proteins to those present in total library. As expected, the % detection decreases as the ion library size increases (Figure 4, top). However the library generation time also increased (Figure 4, bottom). Microflow LC was used during creation of the 2D library, which can help speed up the process of library building and of course the number of fractions and cell lines analyzed used can be varied. A researcher can balance all these considerations when deciding on the right ion library strategy for their biological system of study.
Figure 4. Assessing library detection rates and balancing peptide and protein library size with library generation time. Larger ion libraries allow more peptides and proteins to be identified and quantified, but need more time to be generated from consecutive IDA experiments. (Top) As the size of the ion library increases, the number of peptides and proteins quantified increases. Here the total # of peptides/proteins quantified at <1%FDR and <20% CV (solid bars) are plotted along with the portion of the library that was not quantified (not detected at 1% FDR or failed the stringent quantitation filters, hashed bars). The % detection of the entire library used can then be computed (orange). On the small library, 70-80% of proteins in the library are quantified, whereas only 40% of the protein forms are quantified from the pan human library. (Bottom) Based on the library depth needed, the total library generation time can be balanced by the number of IDA experiments, the amount of fractionation used, the number of related cell types, nanoflow vs microflow LC, etc. Note that the computation of library generation time includes the load, wash and re-equilibration times of the LC system.
Conclusions
High quality SWATH Acquisition can be acquired using Variable Window acquisition and 100 Q1 windows on TripleTOF Systems. These data files contain a tremendous amount of quantitative data and to fully mine the potential information from this data, deep ion libraries are key for targeted data processing. Here, we demonstrate significant gains in peptides and proteins quantified (332% and 118% gains, respectively) when transitioning from a simple 1D ion library to a pan human library. For the data file investigated here, 5922 proteins were quantified at <20% CV when processed using the very comprehensive pan human library. Therefore, in addition to optimizing experimental conditions for SWATH Acquisition, choosing an appropriation ion library strategy that matches the depth of coverage required for the biology under study is also important.
References
- Improved Data Quality Using Variable Q1 Window Widths in SWATH® Acquisition, SCIEX technical note RUO-MKT-02-2879-A.
- Rosenberger G et al. (2014) Scientific Data, 1, 140031.
- Selevsek, N., Chang, C.-Y., Gillet, L.C., Navarro, P., Bernhardt, O.M., Reiter, L., Cheng, L.-Y., Vitek, O., Aebersold, R., (2015) Mol Cell Prot. 14, 739-749.
Related content
- Simplifying the Use of Ion Libraries During Data Processing of SWATH Acquisition Proteomics Data, SCIEX technical note RUO-MKT-02-7514-A.