Methods
Sample preparation: A digested K562 (human immortalized myelogenous leukemia) cell lysate was used for analysis in all experiments (SWATH Performance Kit - P/N 5045757). The sample was dissolved in water with 0.1% formic acid. Protein loads ranging from 25 ng to 2 µg were used in experiments.
Chromatography: A Waters ACQUITY UPLC M-class system was used in a trap-elute workflow. A Phenomenex Kinetex 2.6 µm XB-C18 100 Å, 150 x 0.3 mm LC column was used (P/N 00F-4496-AC) with a Phenomenex micro trap (P/N 05N-4252-AC). The column temperature was 30 ºC. Mobile phase A was water with 0.1% formic acid. Mobile phase B was acetonitrile with 0.1% formic acid. Samples were trapped at 10 µL/min for 3 min at 100% mobile phase A. Four linear gradients (5, 10, 20, and 45 min) were tested using a flow rate of 5 µL/min with the following parameters: 5 and 10 min gradients: 3-35% B, 20 min gradient: 3-32% B, 45 min gradient: 3-30% B.
Mass spectrometry: A SCIEX ZenoTOF 7600 system equipped with the OptiFlow source was used with both the microflow probe and ESI calibration probe.5,8 Source conditions were as follows: CUR, 30 psi; GS1, 10 psi; GS2, 25 psi; IS, 5000 V; TEM, 200 ºC. IDA acquisition parameters that were implemented for all experiments included a TOF MS accumulation time of 100 msec and dynamic collision energy. Only precursors with charge states in the range 2-5 with intensities greater than 100 cps were selected for fragmentation.
As part of the design of experiments (DOE), three acquisition variables were tested: the number of different precursor ions selected for MS/MS for each cycle, the accumulation time for each MS/MS spectrum, and the exclusion time before precursor ions with the same m/z would be reconsidered for MS/MS analysis.
Data processing: Cloud-based data processing was used with the ProteinPilot app in the OneOmics suite.6 All searches were performed as thorough searches, with biological modifications included. Only the resulting proteins and peptides identified at <1% global FDR from fit were used in data analysis. Protein and peptide identification results were visualized and compared in data dashboards in the ProteinPilot app and the FDR (false discovery rate) Comparison Template.7
Finding optimal IDA parameters
In order to optimize the acquisition parameters for IDA, a DOE approach was used, enabling the construction of models to identify optimal parameters. This approach contrasts with a One Factor at a Time (OFAT) approach in that all parameters can be efficiently explored at the same time, instead of varying one parameter at a time and examining the impact on the response. For each condition, the number of MS/MS, the accumulation time and the exclusion time were varied, with three values tested for each. A full factorial design for modeling was used with randomization in sample batches in order to generate data using all possible combinations of factors.
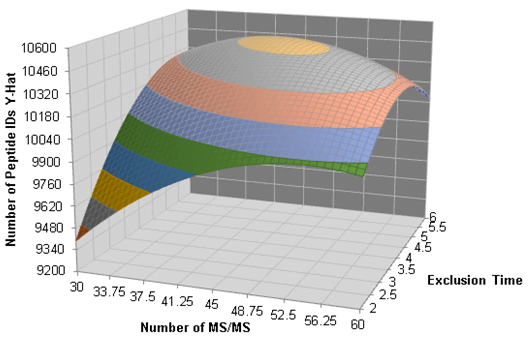
Figure 2 shows an example of a three dimensional surface plot generated from the data acquired during the DOE optimization procedures for a 10 min gradient length. From the shape of the surface, the optimal settings can be determined as well as the size of the sweet spot for optimization. The plot makes it very easy to see that the optimal settings exist somewhere around 45 MS/MS per cycle and an exclusion time of around 4 seconds when running a 10 min gradient. The shape of the 3D surface shows that the number of MS/MS per cycle has a large effect on results with lower numbers of MS/MS and shorter exclusion times resulting in fewer peptide IDs overall.
The other plots indicated the best accumulation time was a very fast acquisition rate of 10 msec per MS/MS (data not shown). The DOE results also included a Pareto analysis that indicated the most important setting to consider was the accumulation time for each MS/MS. The ability to acquire MS/MS at extremely fast acquisition rates (down to 5 msec per spectrum) while maintaining high spectral resolution (>30000 for the higher m/z fragments) is a key advantage of the ZenoTOF 7600 system.
After the DOE experiments provided the best settings for specific sample loadings, further optimization was performed at a variety of other sample loadings for 10 and 45 min gradient lengths. Results were analyzed using the FDR Comparison Template provided in combination with the ProteinPilot app, which facilitates generation of data visuals from protein ID results. In Figure 3, the number of peptide IDs (circles) and protein IDs (diamonds) are plotted as a function of the accumulation time for a 25 ng sample load injected 3x for each experiment using the 10 min gradient. As shown for this data set, the highest numbers of IDs are obtained with slightly longer accumulation times of 20 msec and 60 MS/MS per cycle.
When all experiments were completed, a summary table was compiled of the optimal IDA settings as a function of gradient length (Table 1). As shown for a sample load of 200 ng, the accumulation time optimized at higher values for longer gradient lengths, with MS/MS and exclusion time parameters optimizing in a similar range for all tested gradients.
Sensitivity gains using Zeno MS/MS
Orthogonal injection QTOF instruments typically achieve only 5-25% duty cycle for injection of ions into the TOF region.3 This is largely due to the asynchronous nature of coupling of a continuous ion beam source (electrospray) to a mass analyzer that operates under pulsed conditions (orthogonal TOF). The ZenoTOF 7600 system uses Zeno trap technology to increase the duty cycle to ≥90% across the entire mass range. This results in gains of 4-25 fold in MS/MS sensitivity.3 These duty cycle enhancements are accomplished by trapping ions from the collision cell and gating them into the TOF accelerator region such that all ions arrive at the same time and in a more condensed packet.
Figure 4 shows the impact of activating the Zeno trap on the MS/MS signal for a 400 ng injection of K562 cell lysate and 10 minute LC gradient. Optimum IDA settings were used for this gradient length with 45 MS/MS per cycle, 10 msec accumulation time and 5 second exclusion time. The pink trace shows the MS/MS total ion chromatogram (TIC) obtained with the Zeno trap on while the blue trace shows the Zeno trap off. When using the Zeno trap, a large increase in TIC height is observed. Additionally, quality scores for MS/MS spectra increased greatly as well. Using a score threshold of 95, 87% of scores for the data with the Zeno trap on exceeded this threshold, while only 55% of spectra exceeded this score for the data with the Zeno trap off. The resulting increases in protein and peptide are summarized in Table 2 and Figure 5.
Figure 5 shows the comparison data for Zeno trap on and off for injection of 200 ng K562 cell digest for all gradients tested. Here, the peptide IDs are plotted as a function of the gradient length. While specific gains vary as a function of gradient length, gains of more than 55% are observed for longer gradients as shown in Figure 5 and Table 2.
More peptides and proteins identified
A comparison study was performed for the ZenoTOF 7600 system versus the TripleTOF 6600 system to better understand the gains achieved from the new instrument as a whole. Both short and long gradient conditions were tested as well as many different protein loading amounts with the aim of covering a range of typical laboratory scenarios.
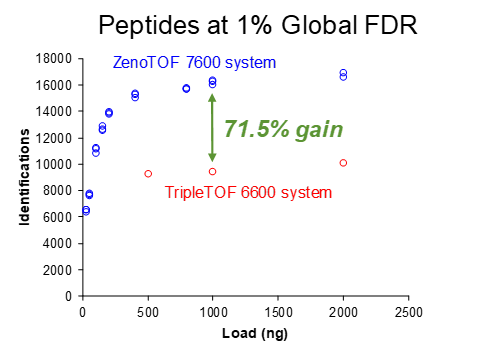
Figure 6 shows the number of proteins identified for replicate protein loadings using a 10-minute gradient. When looking at the low end of the curve, loadings of only 100 ng produce more peptide and protein IDs than loading 5x more (500 ng) on the TripleTOF 6600 system. Table 3 summarizes the large percentage of gains for 1000 ng protein loadings as observed using the ZenoTOF 7600 system.
Figure 7 shows the same experiments, but for a 45-minute gradient, with replicate loadings for the ZenoTOF 7600 system plotted. Again, the ZenoTOF 7600 system produces many more peptide and protein IDs, and the percentage of gains for 1000 ng are summarized in Table 3.
Conclusions
The ZenoTOF 7600 system provides significant gains in peptide and protein identifications for proteomics experiments. These gains come from a combination of the unique Zeno trap technology and the optimization of acquisition parameters for a series of different workflow scenarios.
- Up to ~45% improvements in the number of protein IDs and up to a 145% increase in the number of peptide IDs versus previous state-of-the-art technology for identical sample loadings
- Alternatively, the increased sensitivity can be used to provide very good protein identification results using 5x lower sample loading, when sample amounts are limiting
- The large increase in the number of IDs can be used to improve the quality of libraries generated for SWATH data processing
- As a complete solution using microflow chromatography, the OptiFlow source and cloud-based ProteinPilot App enable a robust, sensitive and streamlined workflow from acquisition through results generation.

 Click to enlarge
Click to enlarge Click to enlarge
Click to enlarge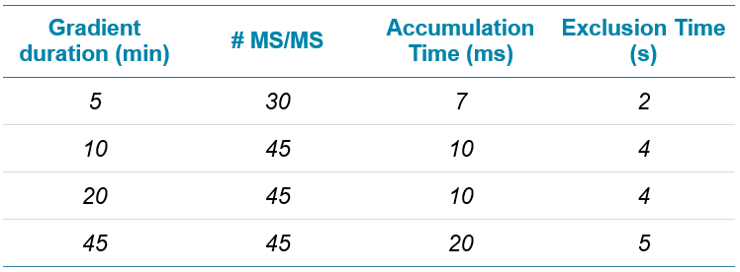 Click to enlarge
Click to enlarge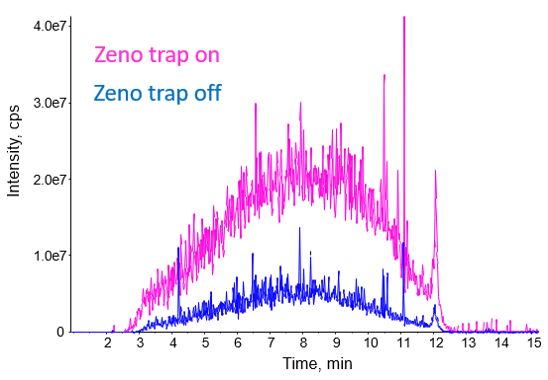 Click to enlarge
Click to enlarge Click to enlarge
Click to enlarge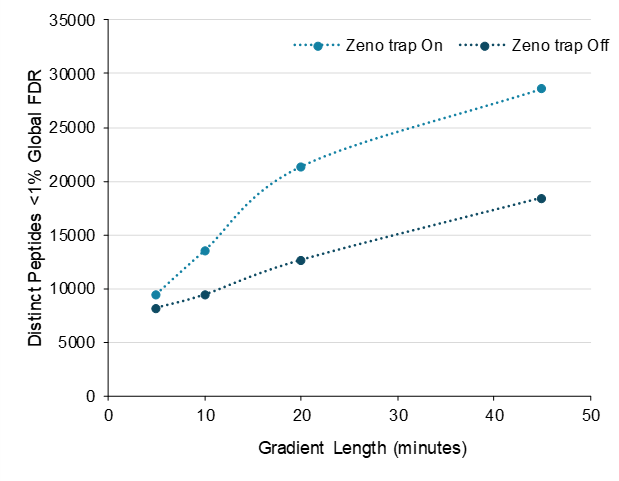 Click to enlarge
Click to enlarge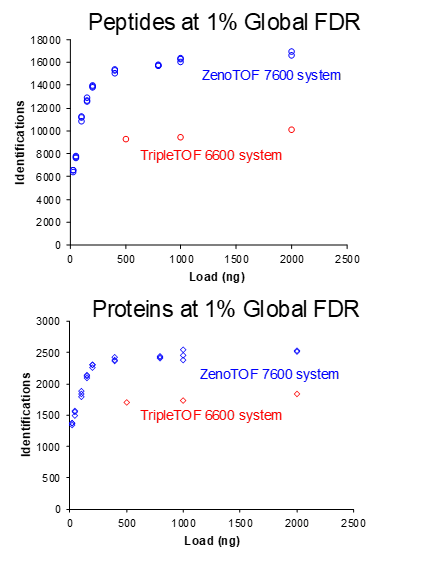 Click to enlarge
Click to enlarge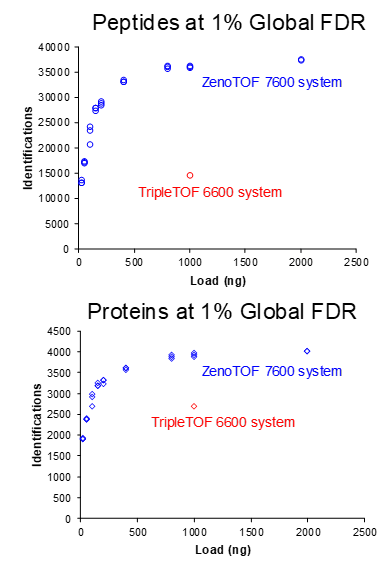 Click to enlarge
Click to enlarge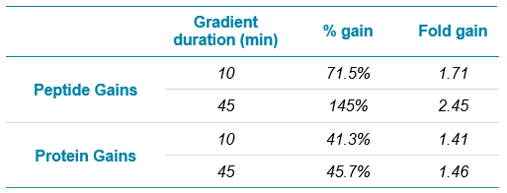 Click to enlarge
Click to enlarge