Evolution of SWATH® Acquisition provides large gains in quantified proteins
Overview of key methodology improvements on TripleTOF® Systems
Christie Hunter1, Nick Morrice2, Joerg Dojahn3
1SCIEX, USA, 2SCIEX, UK, 3SCIEX, Germany
Abstract
The SWATH® Acquisition data independent acquisition strategy has been rapidly adopted by researchers needing robust, comprehensive protein quantitation data for their biological and biomarker research. Significant gains in data quality have been achieved over the last 5 years since launch of the workflow, through innovations in hardware, acquisition methods and library developments. In this work we summarize and quantify the various improvements that have been made to this data independent acquisition (DIA). Comparing to the original implementation of the SWATH method (34fx x 25Da with a small 1D library), very large gains are observed by using a 100 variable window approach with a large ion library, increases of over 100% more proteins and 300% more peptides quantified.

Introduction
The data independent acquisition strategy termed SWATH Acquisition was first described1 and commercialized in 2012. The acquisition strategy used was to step 25 Da Q1 windows across the peptide mass range, collecting high resolution MS/MS data in each step. Then the resulting data was interrogated using a targeted data extraction strategy guided by a small ion library.
Since the first description of the technique, there has been a lot of progress made by the SWATH Acquisition user community in better understanding the best way to collect and process data for large scale protein quantitation experiments. Innovations in data acquisition, improvements in instrument hardware, advances in library generation and large increases in scale of studies have all resulted in large improvements, resulting in the workflow used today.
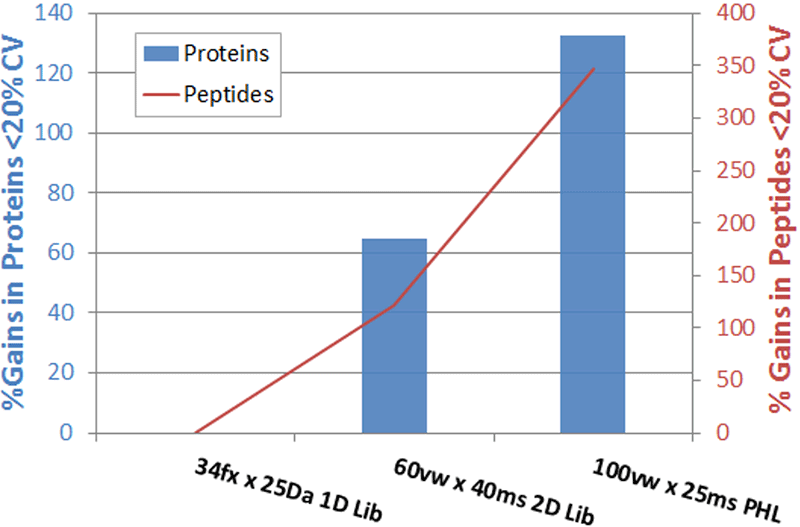
Here the improvements made due to the various method improvements and quantified and summarized. Holding much of the parameters constant (LC, instrument, sample load) and running the various acquisition methods back to back, a good measure of the impact of the techniques can be obtained (Figure 1). Comparing to the method used in 2012 (34 fixed window (fx) x 25Da with a small library), significant gains (>100% protein and 300% peptides quantified) are observed when comparing to results obtained by using a 100 variable window (vw) approach with a large ion library.
Key advances in SWATH® Acquisition
- Variable Q1 Window SWATH Acquisition2
- Specificity of detection and quantitation can be significantly improved through using more, smaller Q1 isolation windows
- By optimizing window width based on m/z density of precursors provides further specificity gains while allowing for broad mass range coverage
- Instrumentation3 – the ability to collect high resolution MS/MS (~30 000 resolution) at high acquisition rates with good dynamic range is key for DIA
- Library generation4,5 – 2 dimensional fractionation for generating deeper ion libraries is important to maximize the quantitative information extracted from SWATH Acquisition data
- Industrialization of workflow through using microflow chromatography provides high robustness and throughput for performing larger scale studies6
Figure 1. Gains in quantified proteins and peptides through SWATH Acquisition innovations. Holding most parameters constant, here the impact of using the original SWATH Acquisition method with a small library is compared with an intermediate approach and today’s best strategy of using 100 variable windows with a large library. Gains of 300% peptides and 120% proteins quantified across replicates are observed.
Methods
Chromatography: Separation of a trypsin digest of human cell lysate was performed on a NanoLC™ 425 System (SCIEX) operating in trap elute mode at microflow rates. A 0.3x150 cm ChromXP™ column (SCIEX) was used with a short gradient (4-32% acetonitrile in 43 min in 0.1% formic acid) at 5 µL/min (total run time 57min6). Total protein digest injected on column was 5 µg.
Mass spectrometry: The MS analysis was performed on multiple TripleTOF® 6600 and 5600 systems (SCIEX), each equipped with Turbo V™ Sources and 25 μm I.D. hybrid electrodes (SCIEX). Variable window SWATH Acquisition methods were built using Analyst® TF Software 1.7. Four different SWATH Acquisition methods were built with different window strategies for comparison (Table 1).
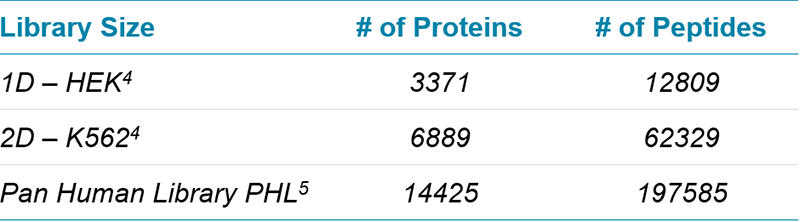
Data processing: Replicate injections of each acquisition condition were processed using SWATH® 2.0 in PeakView® Software 2.2 using 3 different libraries (Table 2). Shared peptides were specifically excluded from quantitation. Results analysis was performed in Excel using the SWATH Acquisition Replicates template7. All protein and peptide numbers reported were determined at <1%FDR and <20% CV across the 5 SWATH acquisition replicates collected.
Table 1. Key parameters in the SWATH Acquisition methods. Four different acquisition methods were used to analyze a cell lysate, to investigate the impact of method on quantitation results. LC gradient, instrument and sample load were held constant.
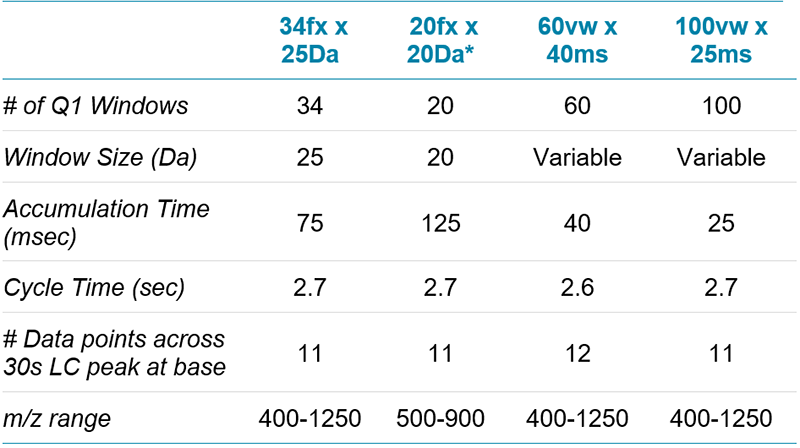
Table 2. SWATH® Acquisition ion libraries for human. A number of ion libraries of increasing size were created for interrogation of SWATH acquisition data (at 1% FDR, excluding modified and shared peptides).
More, smaller windows significantly improve specificity
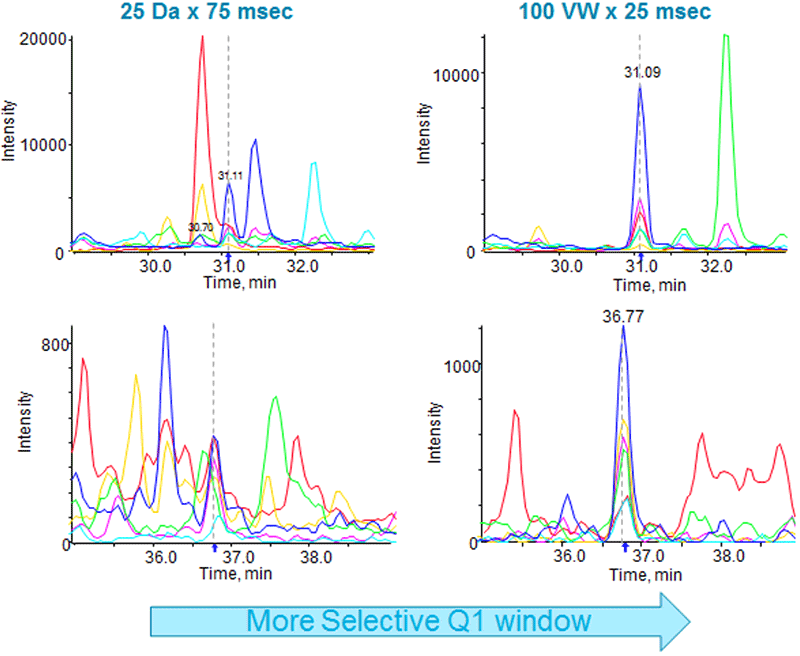
One of the most significant improvements to the SWATH Acquisition strategy is the use of variable sized Q1 isolation windows. By optimizing the size of the window based on the precursor m/z density2, much smaller windows can be used in dense m/z windows, significantly improving the specificity of the data for those targeted peptides. In addition, larger windows can be used across the higher m/z region where far fewer peptide precursors are observed to ensure that proteome coverage is maintained. To test this, we set up a series of experiments on multiple TripleTOF systems where we used increasingly smaller Q1 windows and compared the data to the original 25 Da window strategy (Table 1). After data extraction, we compared the XICs for the same peptide across the different methods to observe the impact on data specificity (Figure 2). There were many cases where significantly improved XIC specificity was observed in the 100 variable window method over the original 25 Da window method.
Figure 2. Decreasing the size of the Q1 isolation windows in SWATH Acquisition improves specificity. In this example, the same sample was analyzed twice on a TripleTOF 6600 System, once with a SWATH Acquisition method using 25 Da wide fixed windows (left, 75 msec accumulation time) and once with a variable window SWATH Acquisition method using 100 windows (right, 25 msec accumulation time). The signal/noise of the detected peptide peak group is greatly improved due to the much smaller Q1 window size used in the variable window method.
Improved peptide quantitation with large number of windows
As expected, this had a large impact on the number of peptides and proteins that could be quantified reproducibly from the replicate datafiles. Comparing just the data using a single large library (PHL, Figure 3, solid bars), there is a ~70-100% gain in the number of peptides quantified between the 25 Da window method (34 windows) and the 60 variable window method. Even more improvements are observed in quantitation (~110-150% peptide increase) when comparing to the 100 variable window strategy. These significant gains are mainly due to the improved S/N obtained on the quantitative XICs on so many of the peptides (Figure 2).
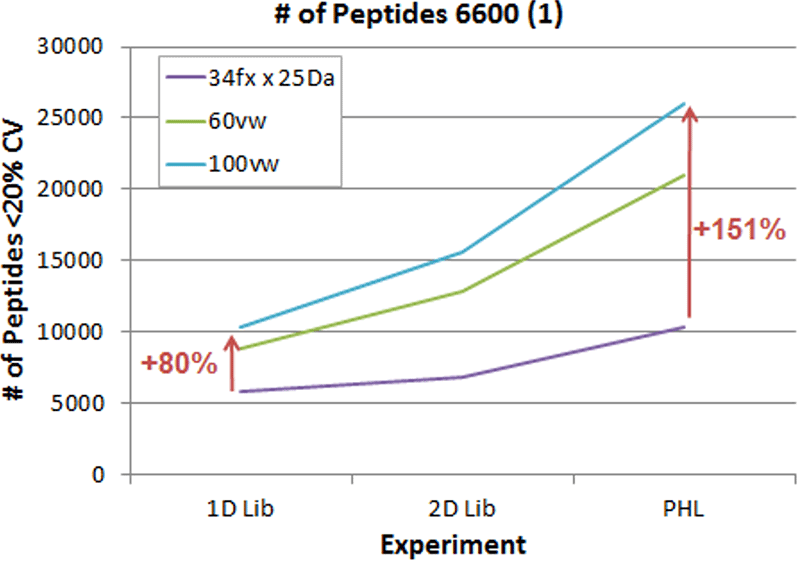
Interestingly, if smaller libraries were used like the 1D library, much smaller gains were seen across the different method (Figure 3, dotted bars). Only 40-50% more peptides were observed in the 60 VW method and ~70-80% more peptides in the 100 VW method over the 25 Da method. To look at this in more detail, a plot of library size vs method was done (Figure 4) and this highlights that when using 60 or 100 VW window SWATH Acquisition methods, it is recommended that larger libraries are also used to fully leverage the amount of quality quantitative data obtained.
The gains were reasonably consistent between the TripleTOF 6600 and 5600 Systems, confirming that the 100 variable window strategy is the optimal strategy for analyzing complex proteomes using SWATH Acquisition.
Figure 3. Gains in peptides quantified using the various acquisition methods and ion libraries. All numbers reported here represent peptides that were detected at <1% FDR and quantified at <20% CV across 5 replicate injections. Data on a HEK cell lysate was collected using the 3 different SWATH acquisition methods (34 fixed 25 Da wide windows, 60 variable windows, and 100 variable windows). Each was processed with the 3 different sized ion libraries and the gains in peptides quantified vs the 25 Da window method is plotted. Numbers of peptides quantified relative to the original implementation of SWATH acquisition (34fx windows with a 1D library) is plotted across all tests. Steady improvements in results are observed as the # of windows increases when using a single library type. In addition, comparing between libraries, gains are also observed as library size gets larger. Overall very large gains in peptides quantified have been made through these workflow innovations.
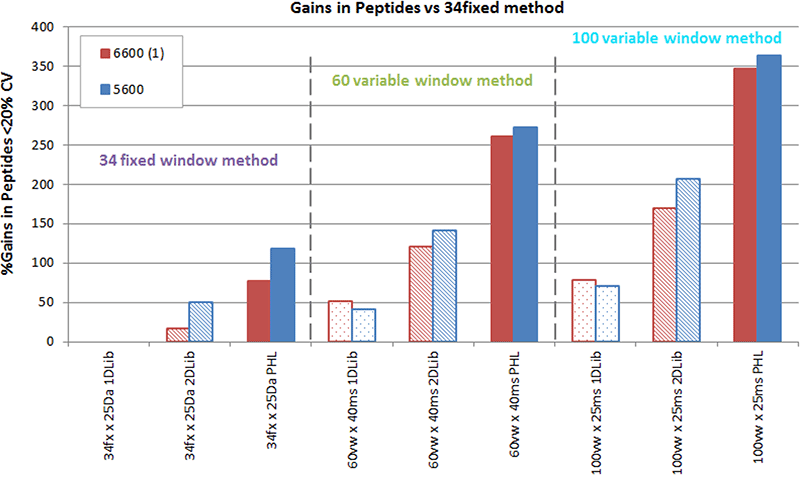
Figure 4. Dependency between library size and SWATH Acquisition strategy. Here the number of peptides quantified for each acquisition strategy was determined using each of the different libraries. This highlights the dependency between using the acquisition method strategy that provides more quantitative information and the use of the larger ion library to fully leverage this added data quality.
SWATH Acquisition in comparison to other DIA strategies
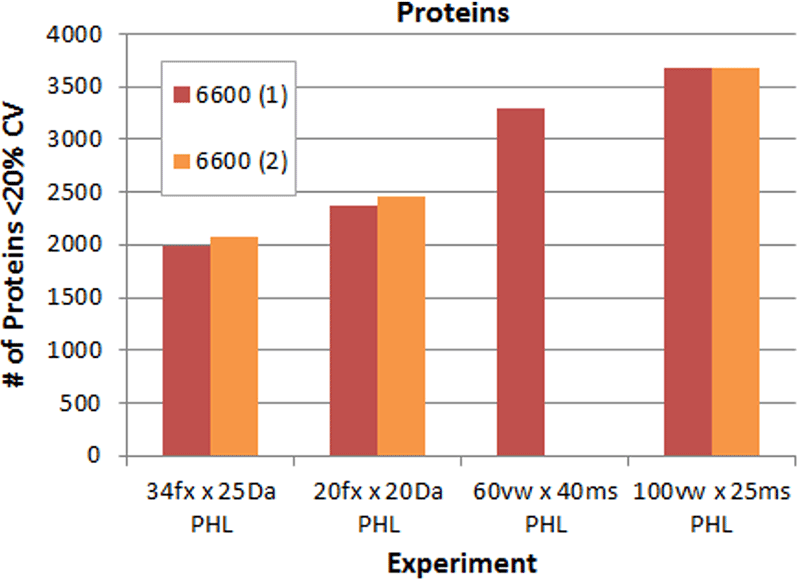
Finally, an additional acquisition method was tested, a method strategy used on other accurate mass systems where high resolution MS/MS acquisition rates are not as high as TripleTOF systems. Here a typical method with 20 fixed windows of 20 Da width is used to interrogate a limited mass range centered around the peptide m/z distribution (500-900m/z). This reduced mass range had to be used, in order to keep the cycle time short to acquire enough data points across the peak for proper quantitation. The concept is that enough peptides are found in the more narrow range to represent most proteins. However in direct comparison, this method was still found to significantly underperform relative to using the 60 or 100 variable window strategy.
Figure 5. Gains in proteins quantified using the four DIA acquisition methods. Comparing at the protein level, again significant gains are observed when the number of windows is increased (decreasing Q1 window size), with over 3500 proteins quantified. Speed of acquiring high resolution MS/MS is a key instrument attribute for performing DIA with large numbers of windows.
Conclusions
The SWATH Acquisition data independent acquisition strategy has been rapidly adopted by researchers needing robust, comprehensive protein quantitation data for their biological and biomarker research.
Significant gains in data quality have been achieved over the last 5 years since launch of the workflow, through innovations in hardware, acquisition methods and library developments. Comparing to the original implementation of the SWATH Acquisition method (34fx x 25Da with a small 1D library), significant gains (>100% protein and 300% peptides quantified) are observed when comparing to results obtained by using a 100 variable window approach with a large ion library.
References
- Gillet LC et al (2012) Mol. Cell. Prot. 11(6), 1-17.
- Improved Data Quality Using Variable Q1 Window Widths in SWATH® Acquisition. SCIEX Technical Note RUO-MKT-02-2879-B.
- TripleTOF® 6600 and 6600+ Systems, SCIEX product page.
- Extending Depth of Coverage with SWATH® Acquisition Using Deeper Ion Libraries, SCIEX technical note RUO-MKT-02-3247-A.
- Rosenberger G et al. (2014) Scientific Data, 1, 140031.
- Microflow SWATH® Acquisition for Industrialized Quantitative Proteomics, SCIEX technical note RUO-MKT-02-3637-A.
- Download SWATH Variable Window Calculator - Excel tool.